

Insights from Mendelian Interferonopathies: Comparison of CANDLE, SAVI with AGS, Monogenic Lupus
- PMID: 27678529
- PMCID: PMC5094849
- DOI: 10.1007/s00109-016-1465-5
Insights from Mendelian Interferonopathies: Comparison of CANDLE, SAVI with AGS, Monogenic Lupus
Abstract
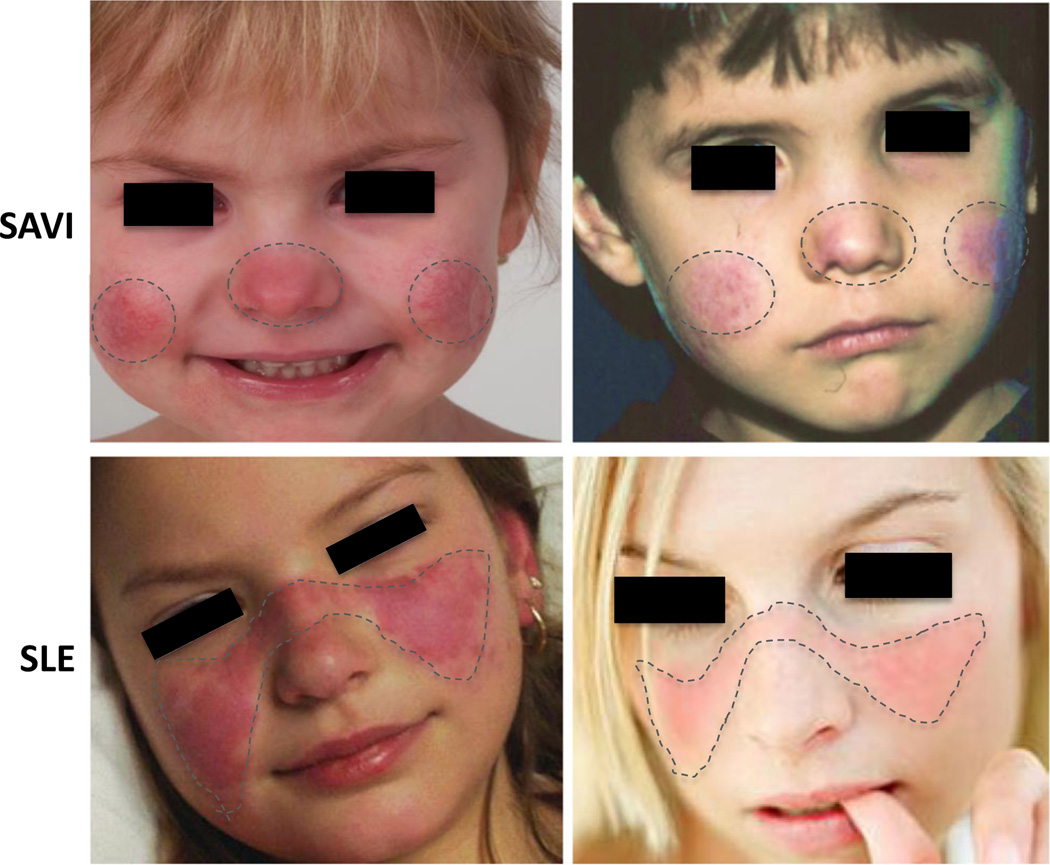
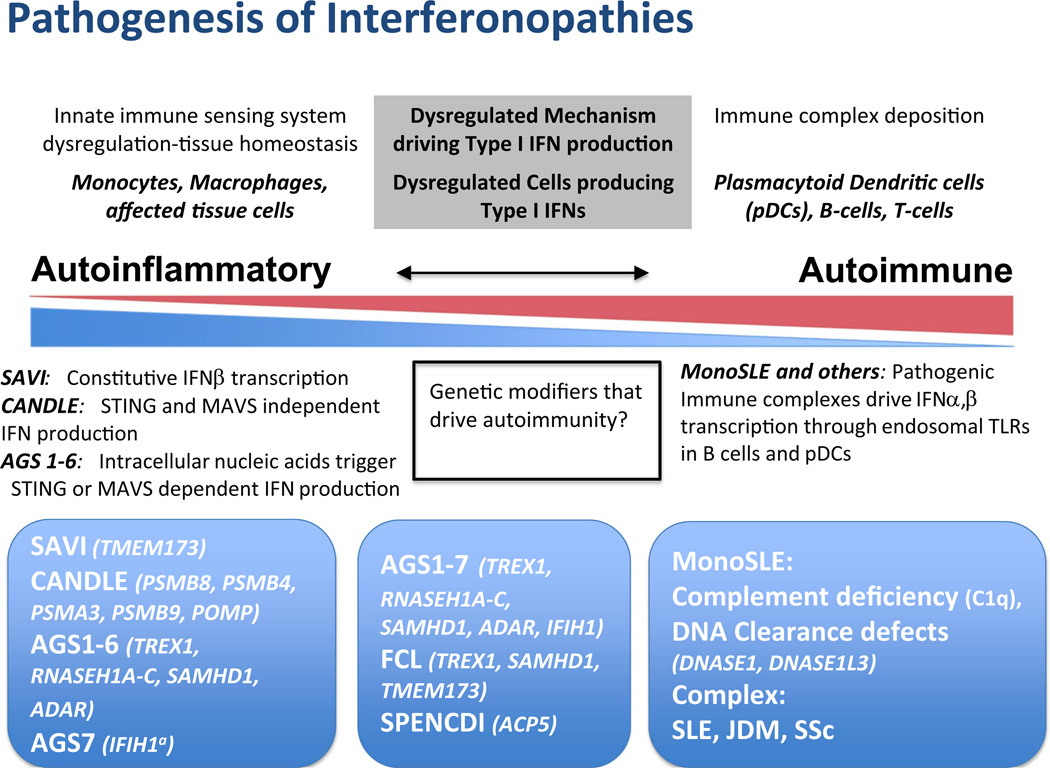
Autoinflammatory disorders are sterile inflammatory conditions characterized by episodes of early-onset fever and disease-specific patterns of organ inflammation. Recently, the discoveries of monogenic disorders with strong type I interferon (IFN) signatures caused by mutations in proteasome degradation and cytoplasmic RNA and DNA sensing pathways suggest a pathogenic role of IFNs in causing autoinflammatory phenotypes. The IFN response gene signature (IGS) has been associated with systemic lupus erythematosus (SLE) and other autoimmune diseases. In this review, we compare the clinical presentations and pathogenesis of two IFN-mediated autoinflammatory diseases, CANDLE and SAVI, with Aicardi Goutières syndrome (AGS) and monogenic forms of SLE (monoSLE) caused by loss-of-function mutations in complement 1 (C1q) or the DNA nucleases, DNASE1 and DNASE1L3. We outline differences in intracellular signaling pathways that fuel a pathologic type I IFN amplification cycle. While IFN amplification is caused by predominantly innate immune cell dysfunction in SAVI, CANDLE, and AGS, autoantibodies to modified RNA and DNA antigens interact with tissues and immune cells including neutrophils and contribute to IFN upregulation in some SLE patients including monoSLE, thus justifying a grouping of "autoinflammatory" and "autoimmune" interferonopathies. Understanding of the differences in the cellular sources and signaling pathways will guide new drug development and the use of emerging targeted therapies.
Keywords: Autoimmune; Autoinflammatory; Candle; Interferonopathies; SAVI; Type I IFN.
Figures
References
-
- Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, Kim PW, Sheikh A, Lee CC, Chen Y, Vera A, et al. Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheumatism. 2012;64:895–907. - PMC - PubMed
-
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12:1159–1169. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
