

De novo phasing with X-ray laser reveals mosquito larvicide BinAB structure
- PMID: 27680699
- PMCID: PMC5161637
- DOI: 10.1038/nature19825
De novo phasing with X-ray laser reveals mosquito larvicide BinAB structure
Abstract
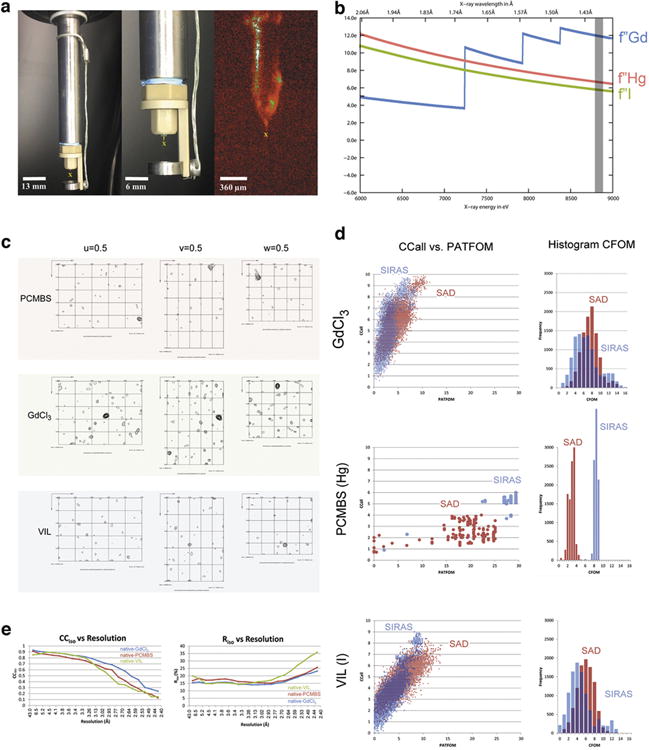
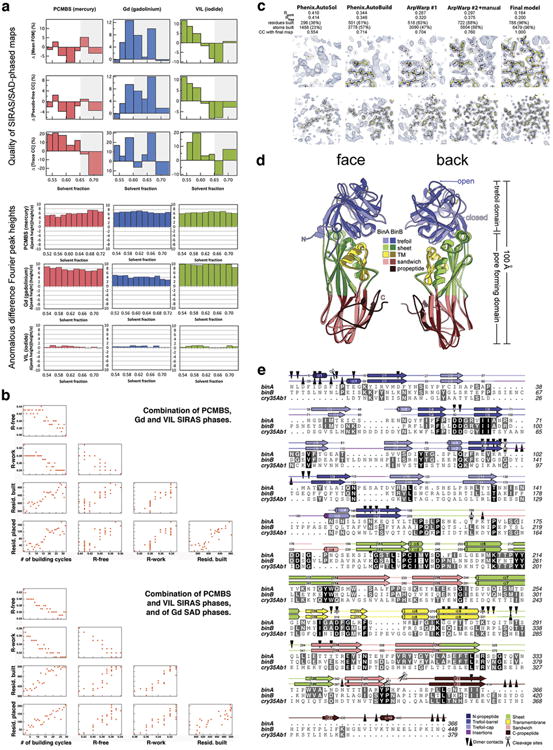
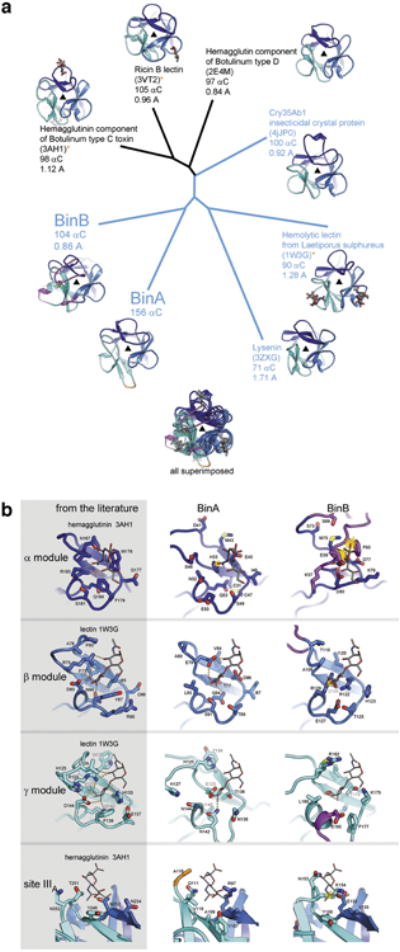
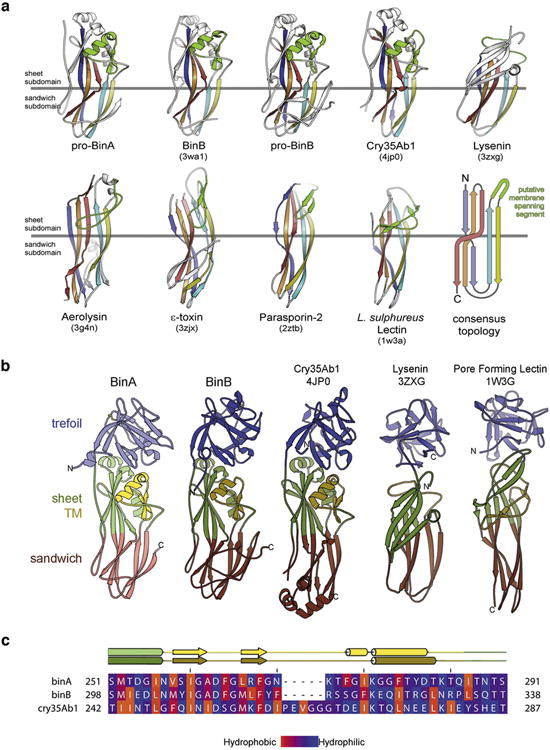
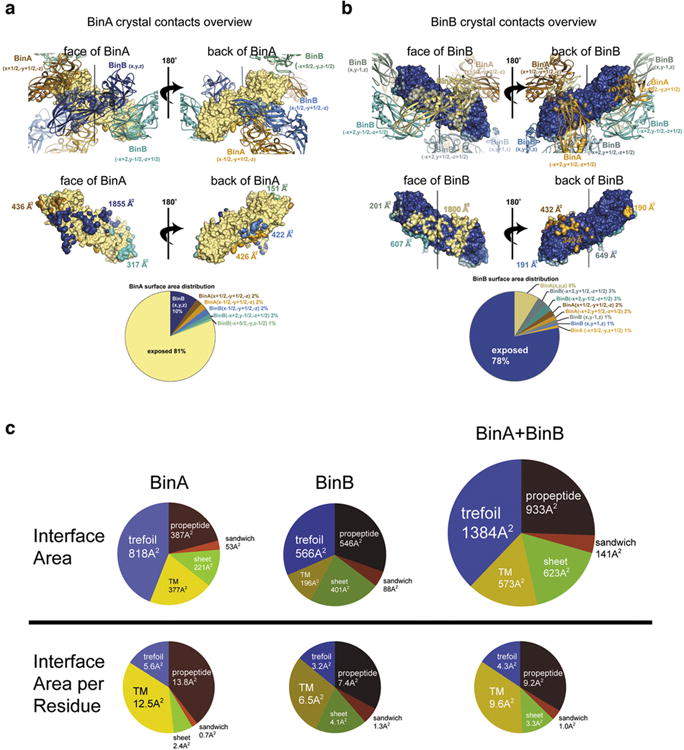
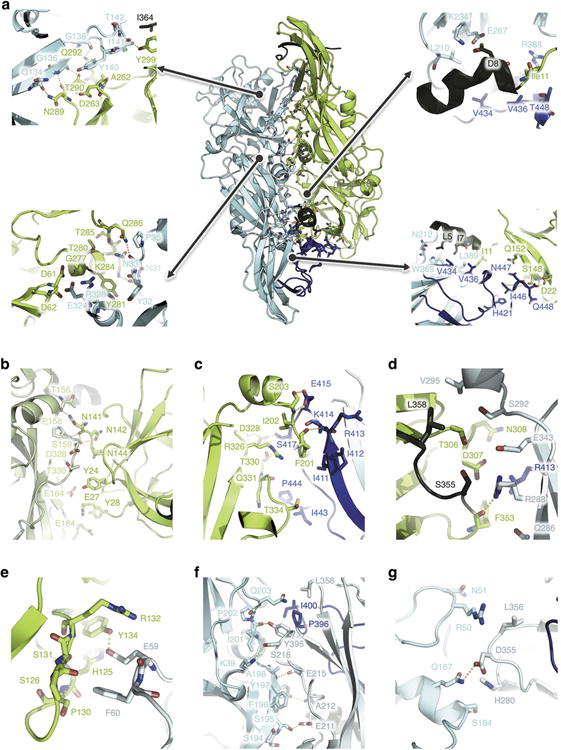
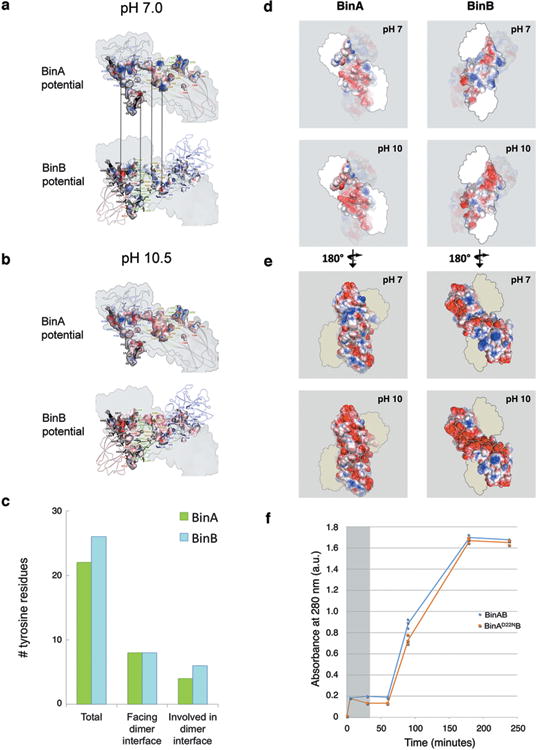
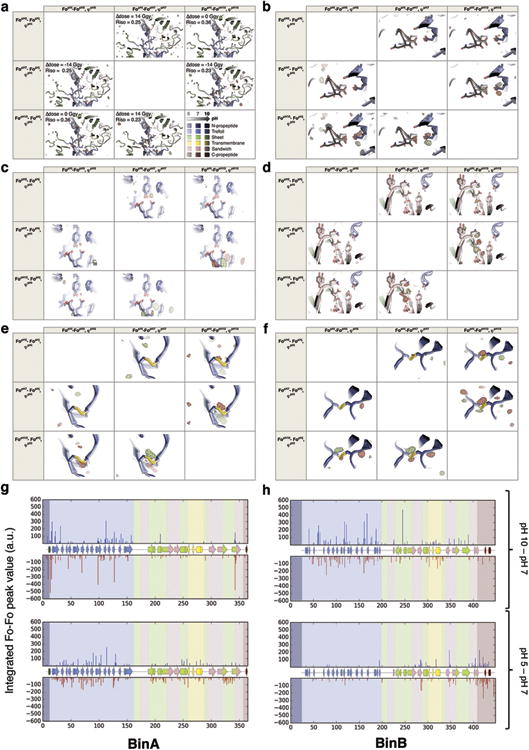
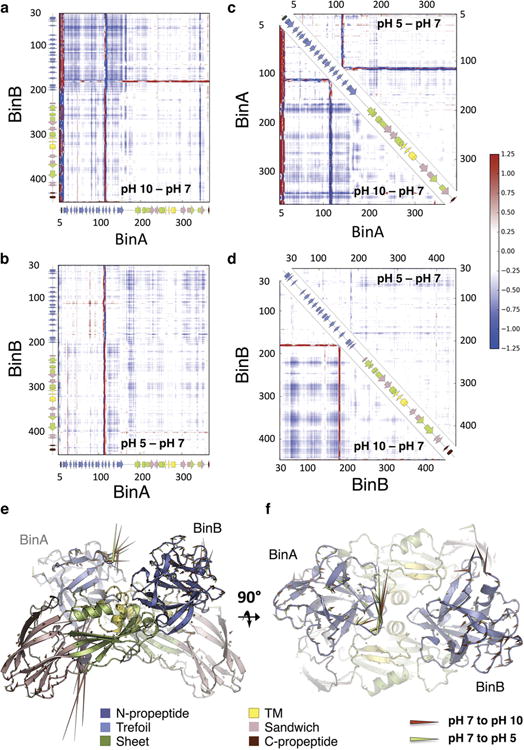
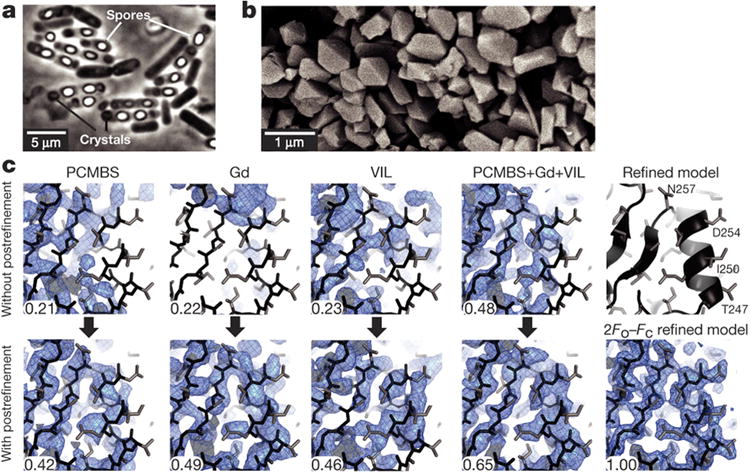
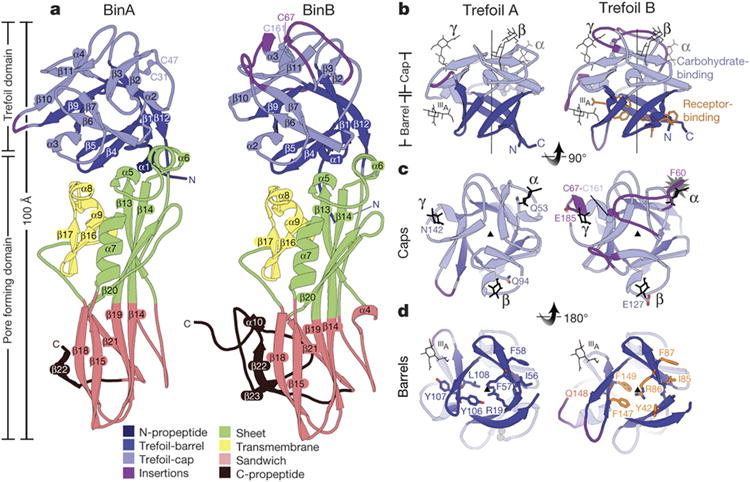
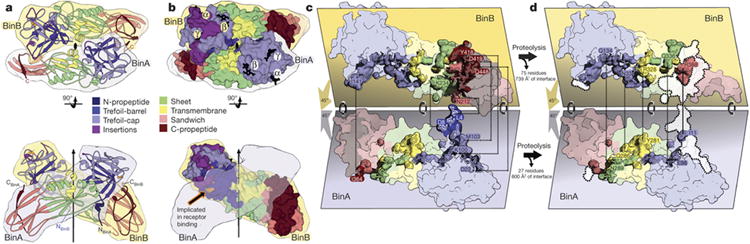
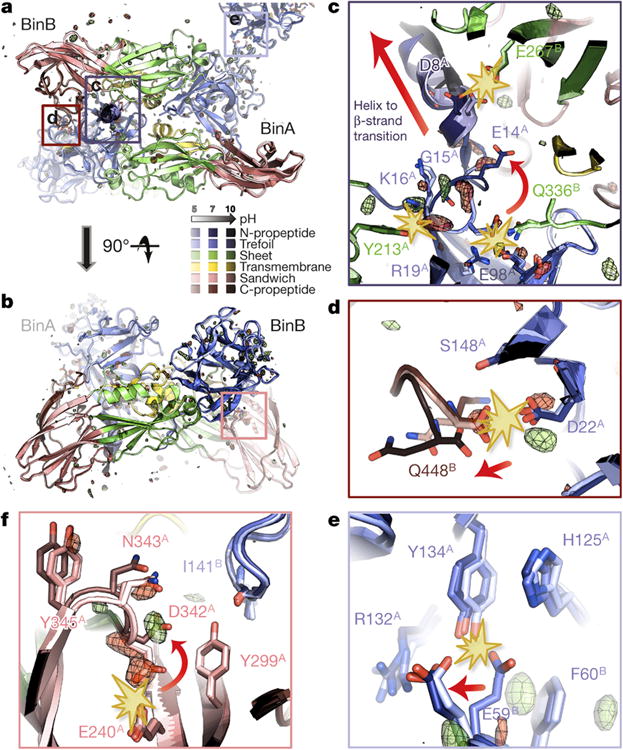
BinAB is a naturally occurring paracrystalline larvicide distributed worldwide to combat the devastating diseases borne by mosquitoes. These crystals are composed of homologous molecules, BinA and BinB, which play distinct roles in the multi-step intoxication process, transforming from harmless, robust crystals, to soluble protoxin heterodimers, to internalized mature toxin, and finally to toxic oligomeric pores. The small size of the crystals-50 unit cells per edge, on average-has impeded structural characterization by conventional means. Here we report the structure of Lysinibacillus sphaericus BinAB solved de novo by serial-femtosecond crystallography at an X-ray free-electron laser. The structure reveals tyrosine- and carboxylate-mediated contacts acting as pH switches to release soluble protoxin in the alkaline larval midgut. An enormous heterodimeric interface appears to be responsible for anchoring BinA to receptor-bound BinB for co-internalization. Remarkably, this interface is largely composed of propeptides, suggesting that proteolytic maturation would trigger dissociation of the heterodimer and progression to pore formation.
Figures
References
-
- Moreira LA, et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell. 2009;139:1268–1278. - PubMed
-
- Alphey N, Bonsall MB, Alphey L. Combining pest control and resistance management: synergy of engineered insects with Bt crops. J Econ Entomol. 2009;102:717–732. - PubMed
-
- Federici BA, Park HW, Bideshi DK. Overview of the basic biology of Bacillus thuringiensis with emphasis on genetic engineering of bacterial larvicides for mosquito control. Open Toxinol J. 2010;3:83–100.
Additional references for Methods, Supplementary Discussion and Supplementary Tables
-
- Park HW, et al. Recombinant larvicidal bacteria with markedly improved efficacy against culex vectors of west nile virus. Am J Trop Med Hyg. 2005;72:732–738. - PubMed
-
- Park HW, Hice RH, Federici BA. Effect of Promoters and Plasmid Copy Number on Cyt1A Synthesis and Crystal Assembly in Bacillus thuringiensis. Curr Microbiol. 2016;72:33–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
