

XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer
- PMID: 27680702
- PMCID: PMC5161658
- DOI: 10.1038/nature19771
XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer
Abstract
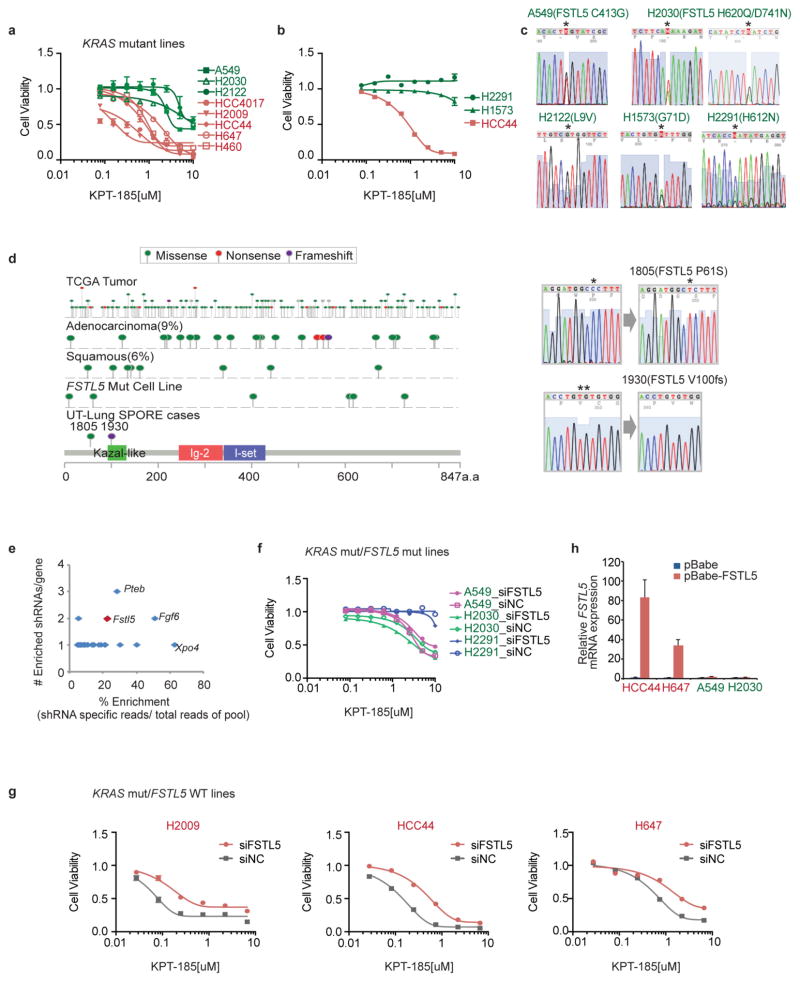
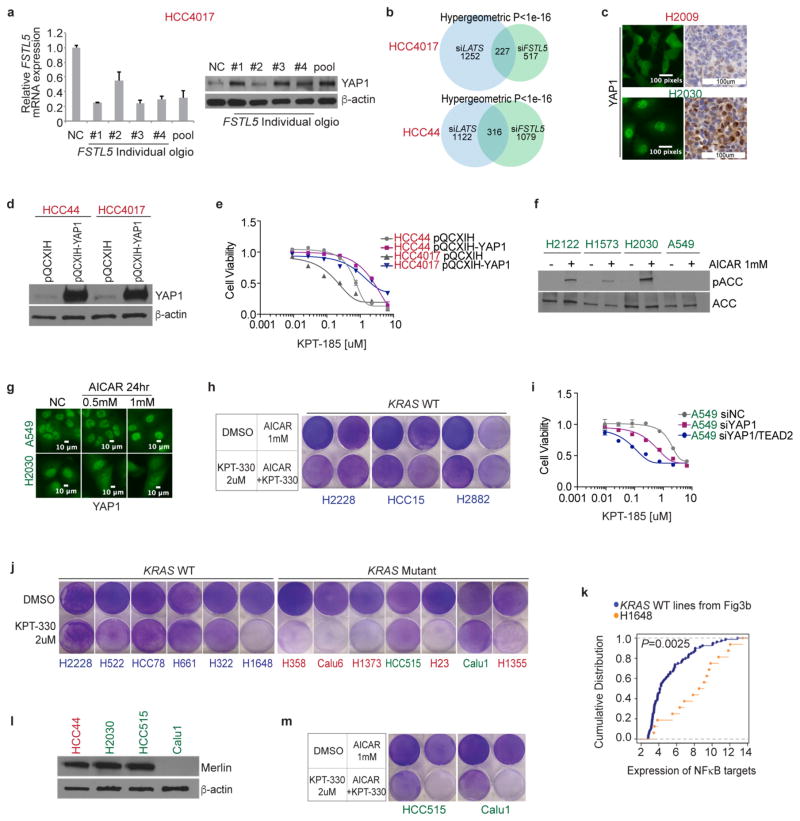
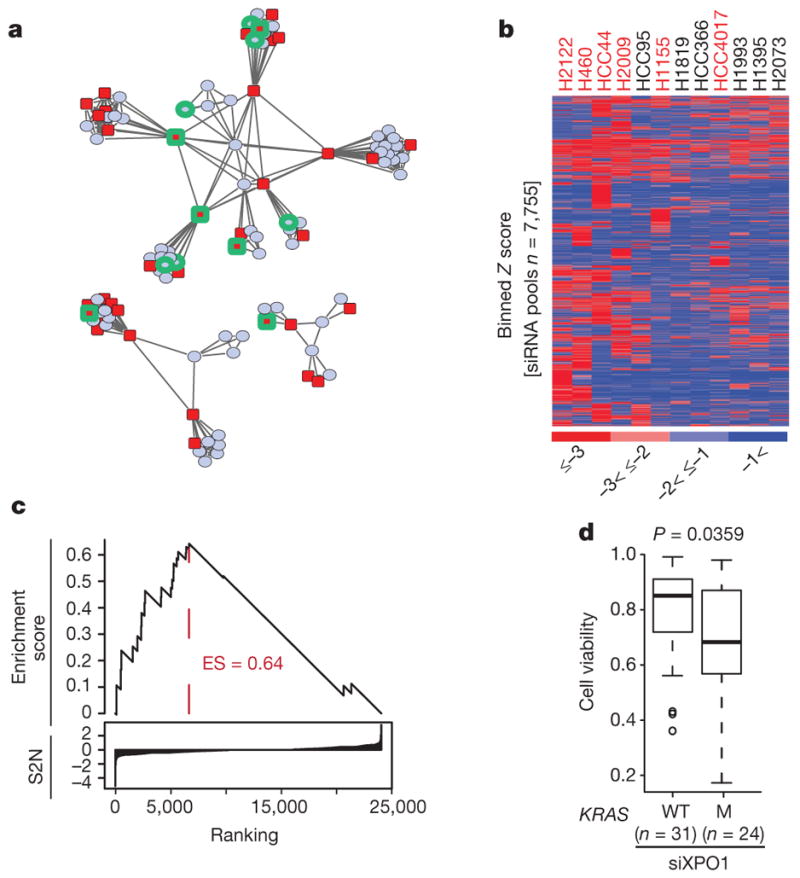
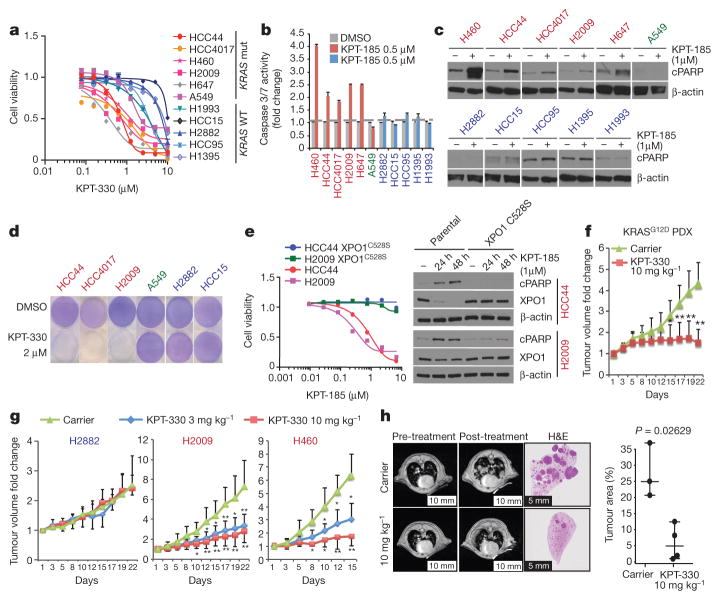
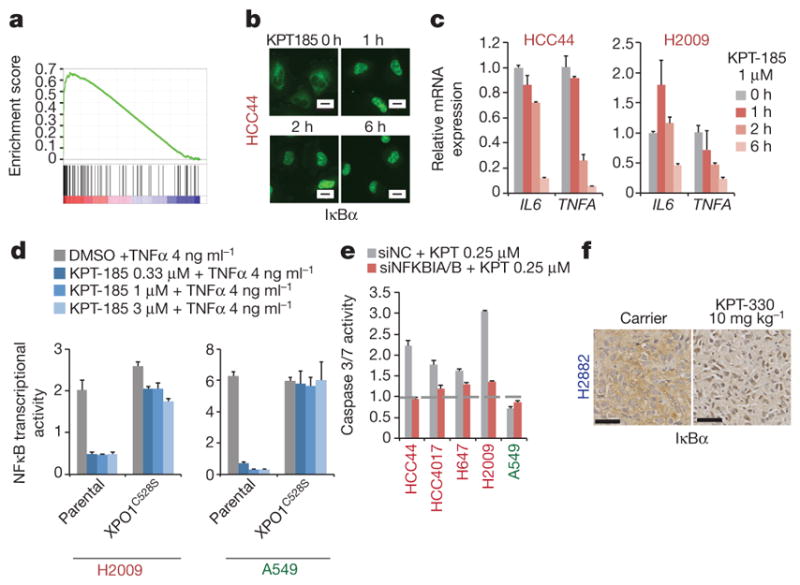
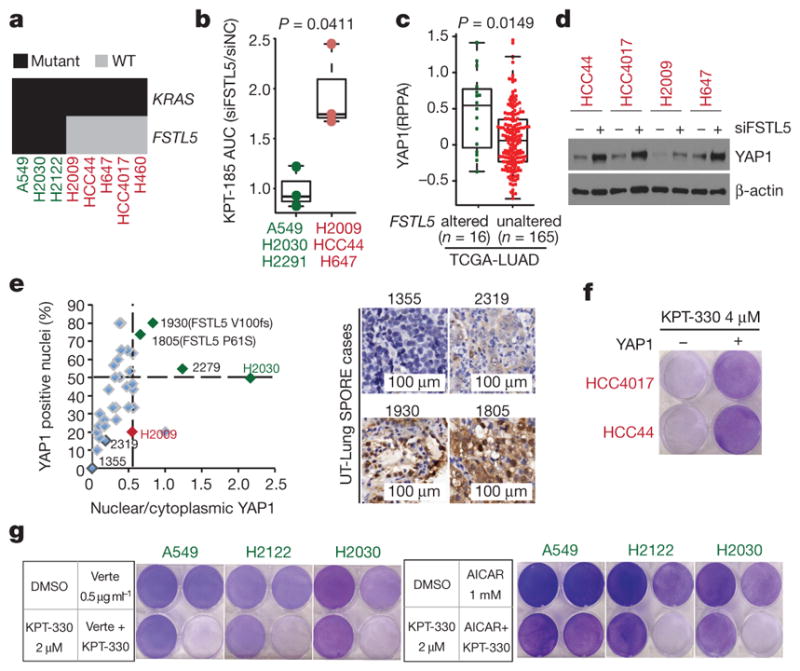
The common participation of oncogenic KRAS proteins in many of the most lethal human cancers, together with the ease of detecting somatic KRAS mutant alleles in patient samples, has spurred persistent and intensive efforts to develop drugs that inhibit KRAS activity. However, advances have been hindered by the pervasive inter- and intra-lineage diversity in the targetable mechanisms that underlie KRAS-driven cancers, limited pharmacological accessibility of many candidate synthetic-lethal interactions and the swift emergence of unanticipated resistance mechanisms to otherwise effective targeted therapies. Here we demonstrate the acute and specific cell-autonomous addiction of KRAS-mutant non-small-cell lung cancer cells to receptor-dependent nuclear export. A multi-genomic, data-driven approach, utilizing 106 human non-small-cell lung cancer cell lines, was used to interrogate 4,725 biological processes with 39,760 short interfering RNA pools for those selectively required for the survival of KRAS-mutant cells that harbour a broad spectrum of phenotypic variation. Nuclear transport machinery was the sole process-level discriminator of statistical significance. Chemical perturbation of the nuclear export receptor XPO1 (also known as CRM1), with a clinically available drug, revealed a robust synthetic-lethal interaction with native or engineered oncogenic KRAS both in vitro and in vivo. The primary mechanism underpinning XPO1 inhibitor sensitivity was intolerance to the accumulation of nuclear IκBα (also known as NFKBIA), with consequent inhibition of NFκB transcription factor activity. Intrinsic resistance associated with concurrent FSTL5 mutations was detected and determined to be a consequence of YAP1 activation via a previously unappreciated FSTL5-Hippo pathway regulatory axis. This occurs in approximately 17% of KRAS-mutant lung cancers, and can be overcome with the co-administration of a YAP1-TEAD inhibitor. These findings indicate that clinically available XPO1 inhibitors are a promising therapeutic strategy for a considerable cohort of patients with lung cancer when coupled to genomics-guided patient selection and observation.
Conflict of interest statement
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
Figures
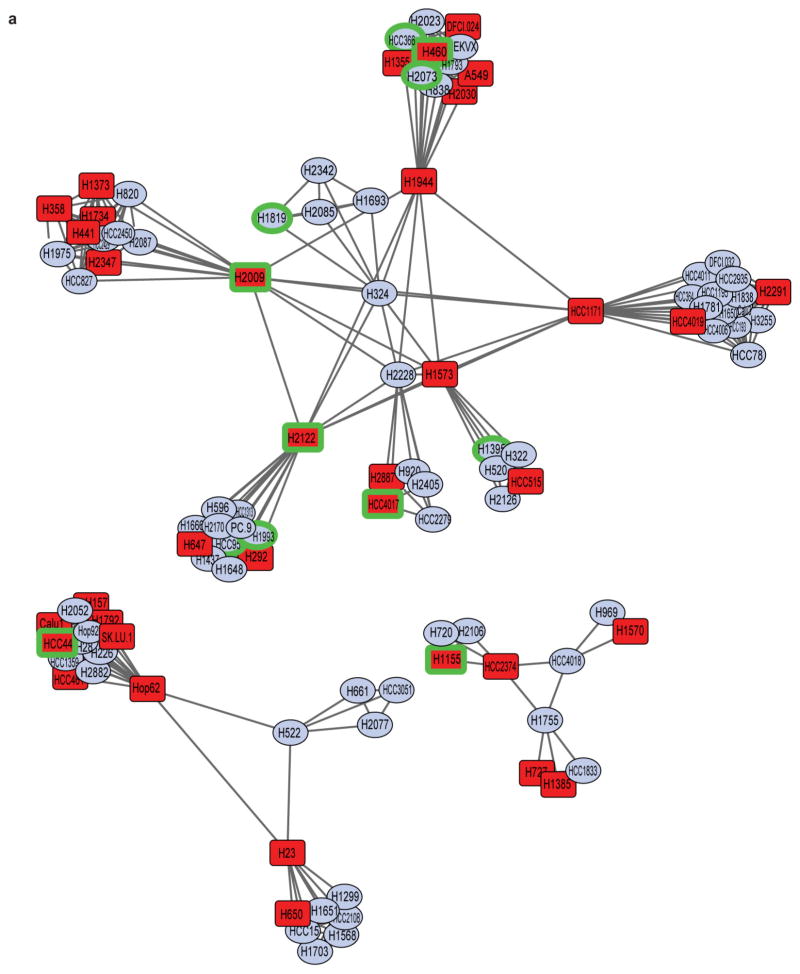
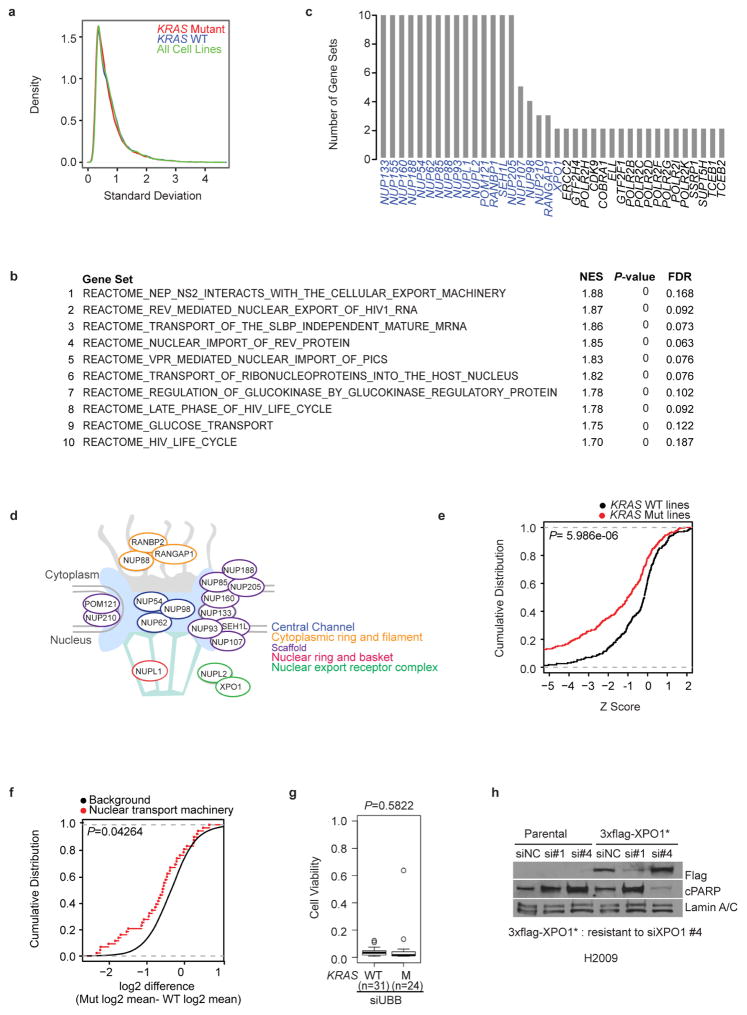
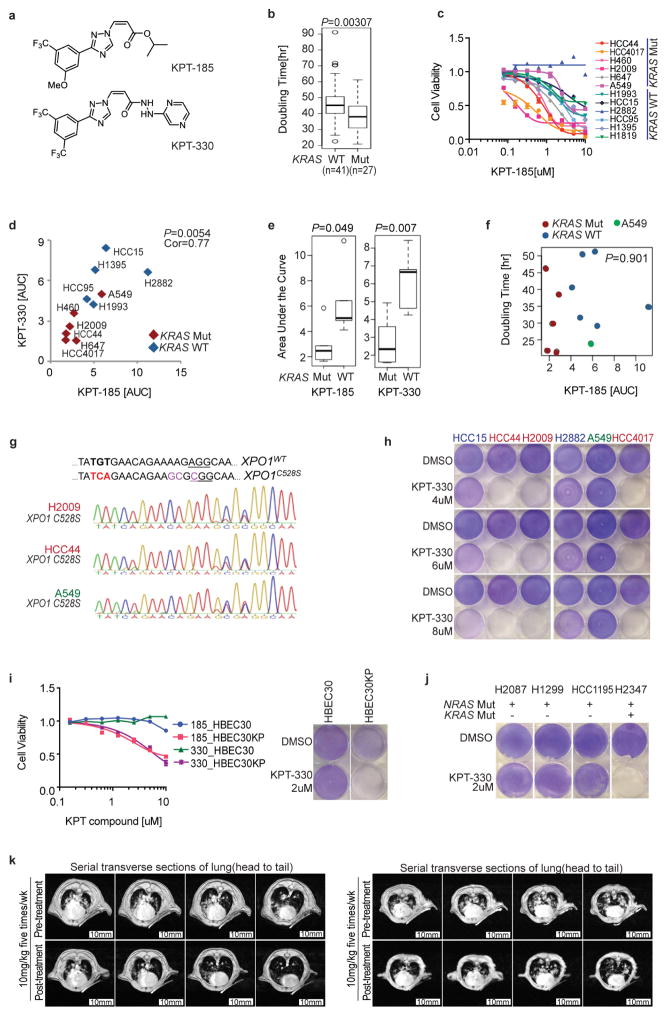
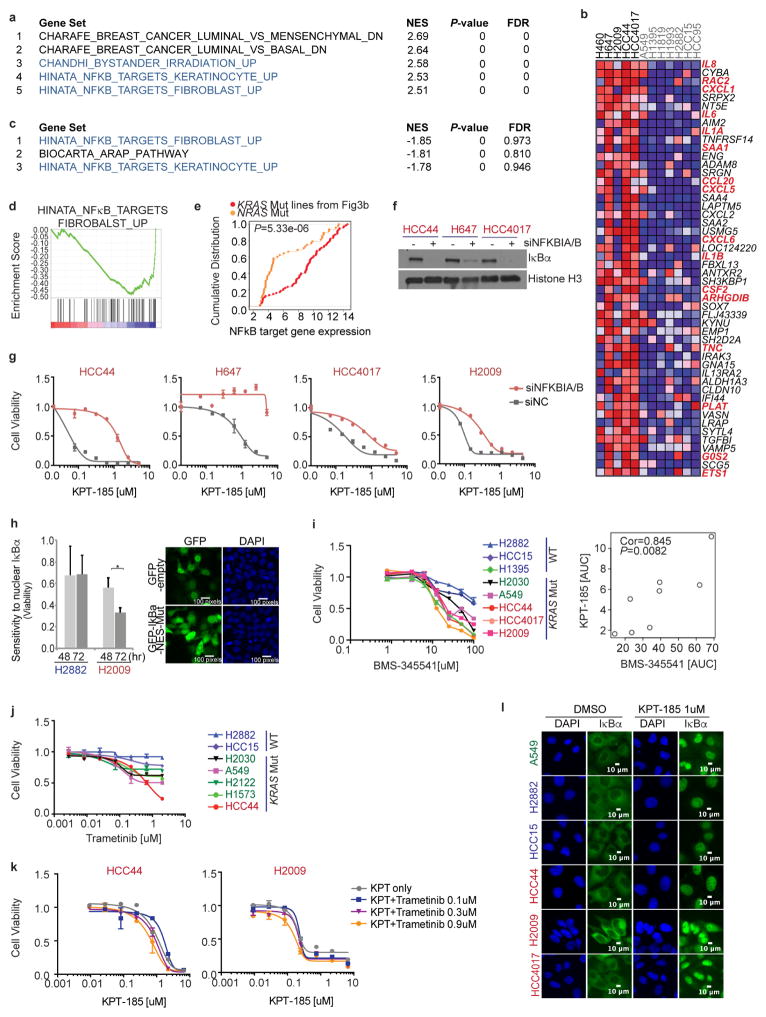
Comment in
-
Exportin 1/chromosome region maintenance 1 as a therapeutic target for lung cancer.Transl Cancer Res. 2017 Feb;6(Suppl 1):S83-S86. doi: 10.21037/tcr.2017.02.35. Transl Cancer Res. 2017. PMID: 30613477 Free PMC article. No abstract available.
References
-
- Kumar MS, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149:642–655. - PubMed
-
- Scholl C, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–834. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
