

Posttranslational arginylation enzyme Ate1 affects DNA mutagenesis by regulating stress response
- PMID: 27685622
- PMCID: PMC5059882
- DOI: 10.1038/cddis.2016.284
Posttranslational arginylation enzyme Ate1 affects DNA mutagenesis by regulating stress response
Abstract
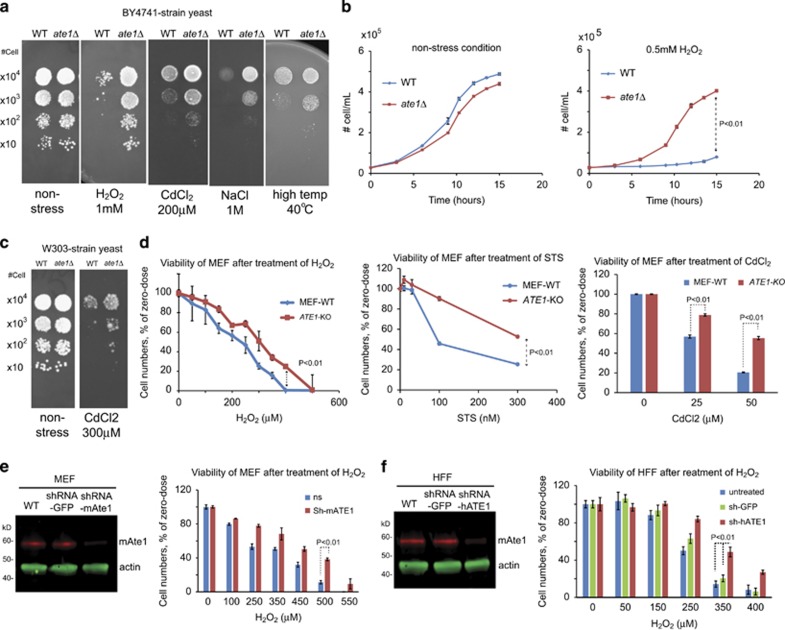
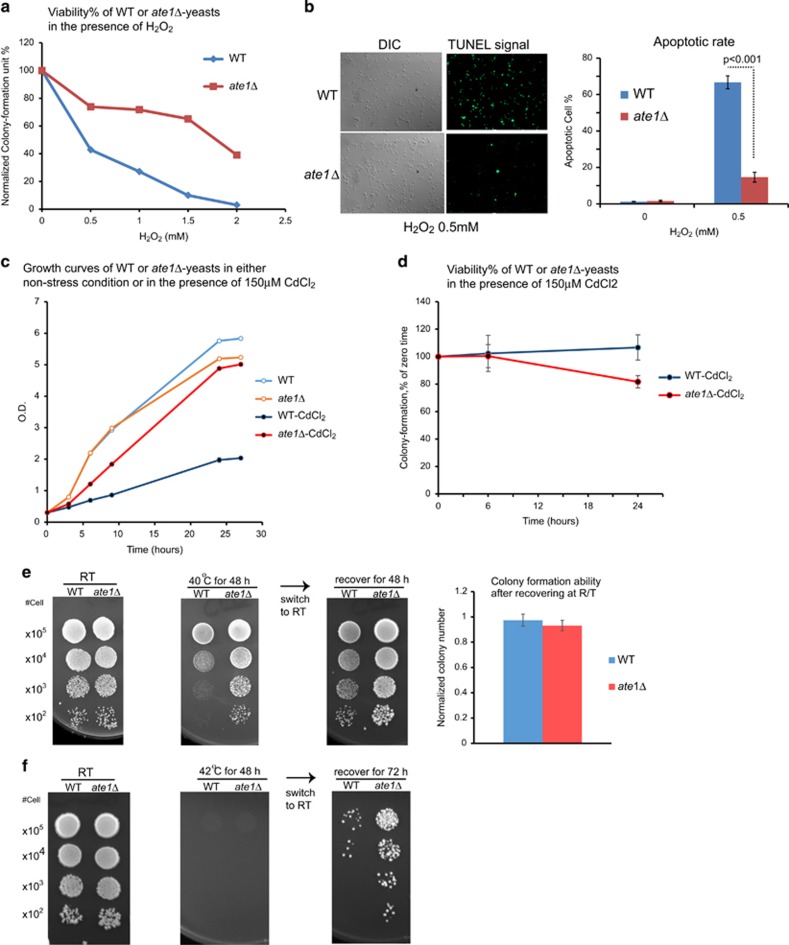
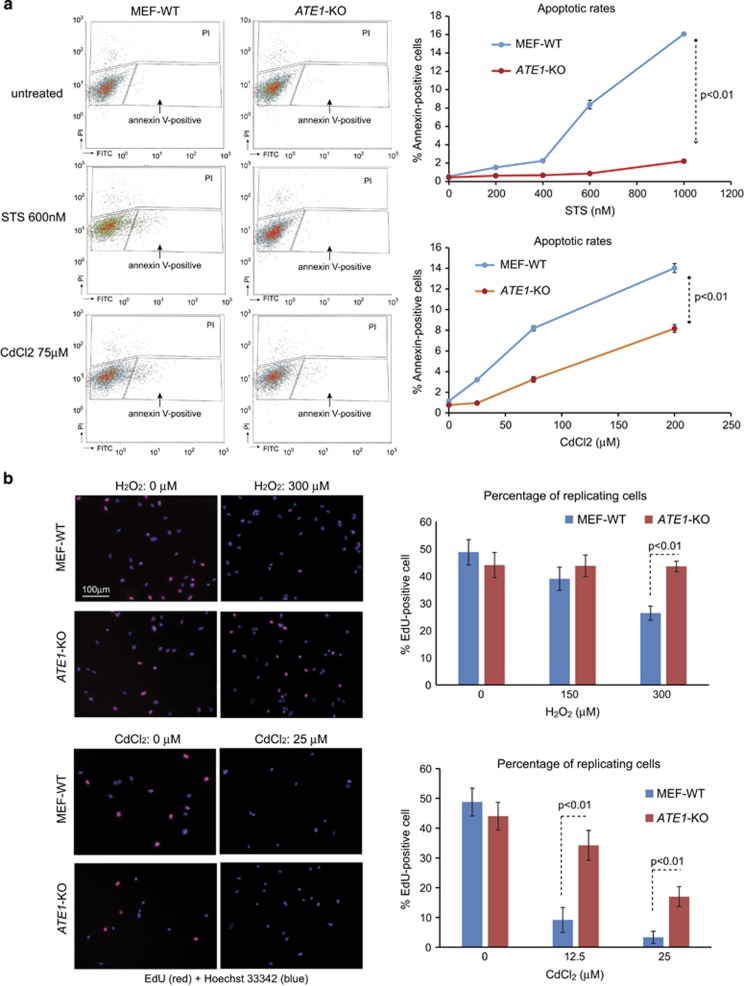
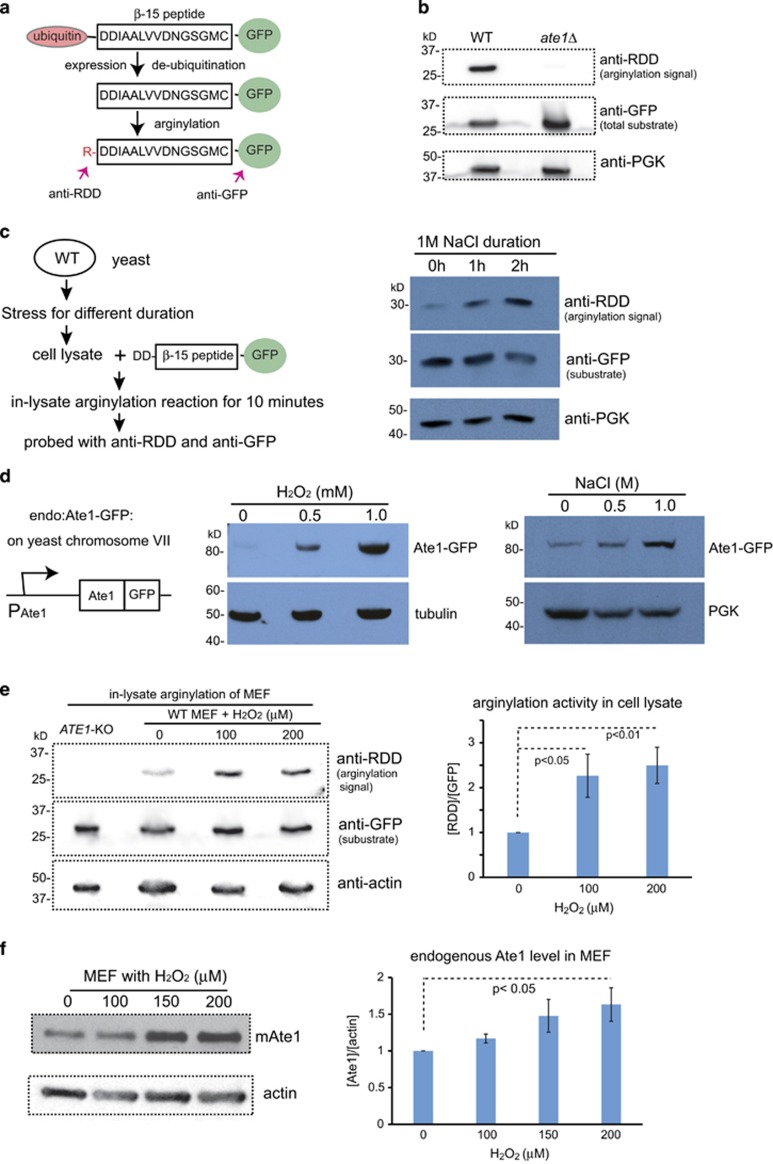
Arginyltransferase 1 (Ate1) mediates protein arginylation, a poorly understood protein posttranslational modification (PTM) in eukaryotic cells. Previous evidence suggest a potential involvement of arginylation in stress response and this PTM was traditionally considered anti-apoptotic based on the studies of individual substrates. However, here we found that arginylation promotes cell death and/or growth arrest, depending on the nature and intensity of the stressing factor. Specifically, in yeast, mouse and human cells, deletion or downregulation of the ATE1 gene disrupts typical stress responses by bypassing growth arrest and suppressing cell death events in the presence of disease-related stressing factors, including oxidative, heat, and osmotic stresses, as well as the exposure to heavy metals or radiation. Conversely, in wild-type cells responding to stress, there is an increase of cellular Ate1 protein level and arginylation activity. Furthermore, the increase of Ate1 protein directly promotes cell death in a manner dependent on its arginylation activity. Finally, we found Ate1 to be required to suppress mutation frequency in yeast and mammalian cells during DNA-damaging conditions such as ultraviolet irradiation. Our study clarifies the role of Ate1/arginylation in stress response and provides a new mechanism to explain the link between Ate1 and a variety of diseases including cancer. This is also the first example that the modulation of the global level of a PTM is capable of affecting DNA mutagenesis.
Figures
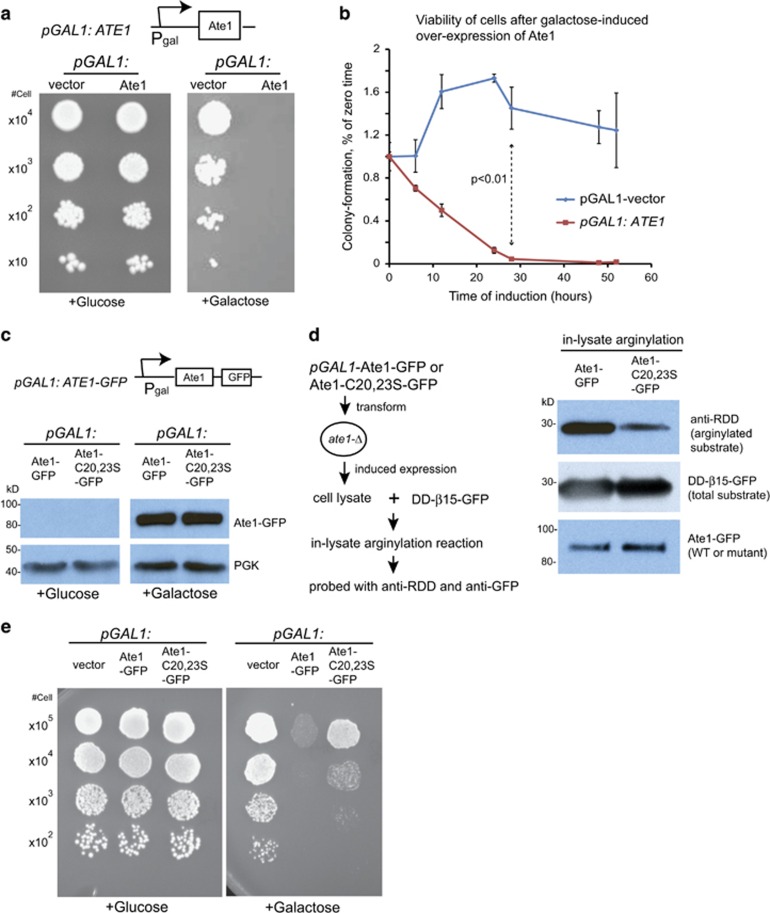
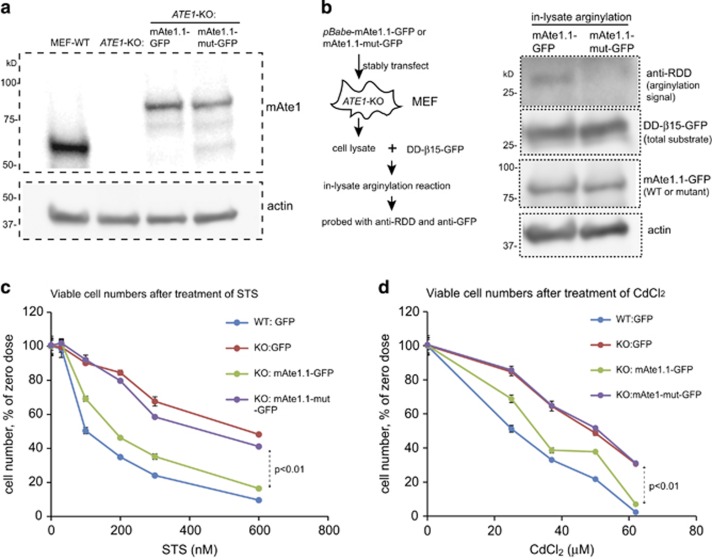
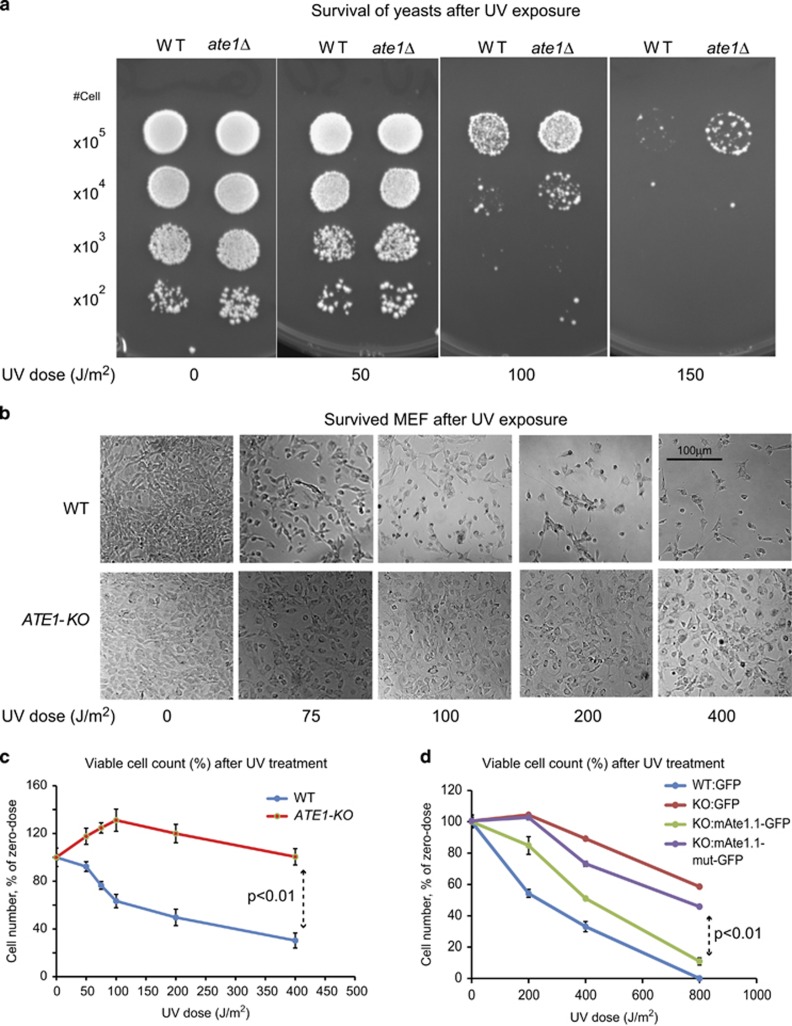
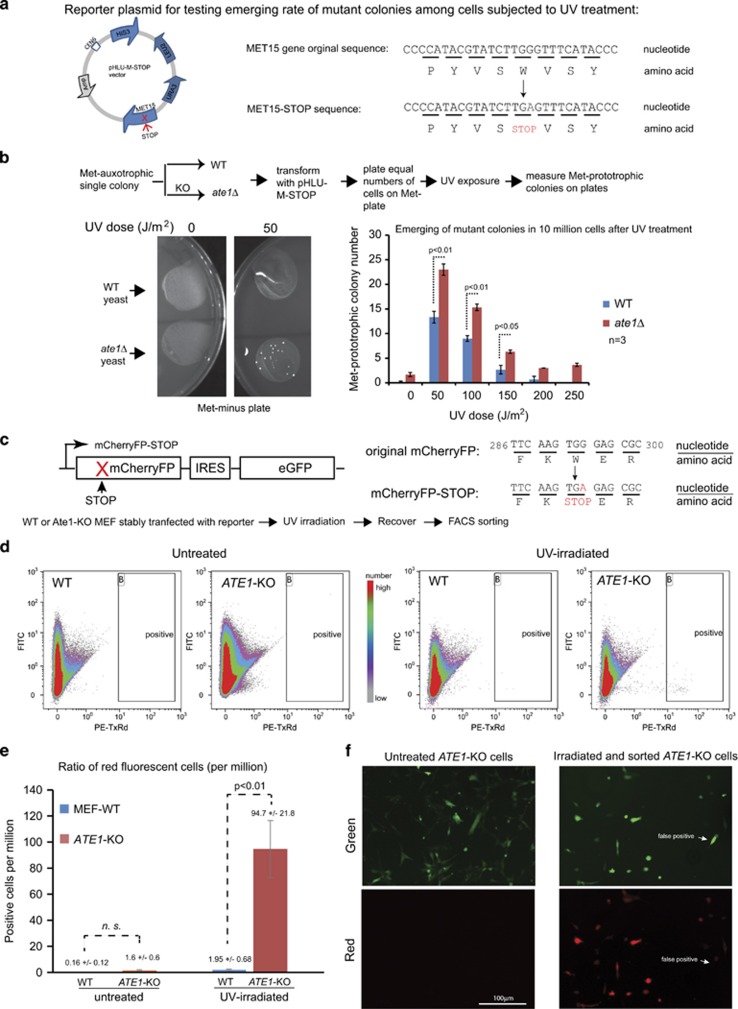
Similar articles
-
Ate1-mediated posttranslational arginylation affects substrate adhesion and cell migration in Dictyostelium discoideum.Mol Biol Cell. 2019 Feb 15;30(4):453-466. doi: 10.1091/mbc.E18-02-0132. Epub 2018 Dec 26. Mol Biol Cell. 2019. PMID: 30586322 Free PMC article.
-
tRNAArg-Derived Fragments Can Serve as Arginine Donors for Protein Arginylation.Cell Chem Biol. 2020 Jul 16;27(7):839-849.e4. doi: 10.1016/j.chembiol.2020.05.013. Epub 2020 Jun 16. Cell Chem Biol. 2020. PMID: 32553119 Free PMC article.
-
Post-translational protein arginylation in the normal nervous system and in neurodegeneration.J Neurochem. 2016 Aug;138(4):506-17. doi: 10.1111/jnc.13708. Epub 2016 Jul 5. J Neurochem. 2016. PMID: 27318192 Review.
-
The preparation of recombinant arginyltransferase 1 (ATE1) for biophysical characterization.Methods Enzymol. 2023;679:235-254. doi: 10.1016/bs.mie.2022.07.036. Epub 2022 Aug 31. Methods Enzymol. 2023. PMID: 36682863 Free PMC article.
-
Posttranslational arginylation as a global biological regulator.Dev Biol. 2011 Oct 1;358(1):1-8. doi: 10.1016/j.ydbio.2011.06.043. Epub 2011 Jul 18. Dev Biol. 2011. PMID: 21784066 Free PMC article. Review.
Cited by
-
Regulating Apoptosis by Degradation: The N-End Rule-Mediated Regulation of Apoptotic Proteolytic Fragments in Mammalian Cells.Int J Mol Sci. 2018 Oct 31;19(11):3414. doi: 10.3390/ijms19113414. Int J Mol Sci. 2018. PMID: 30384441 Free PMC article. Review.
-
Regulation of Mitochondrial Respiratory Chain Complex Levels, Organization, and Function by Arginyltransferase 1.Front Cell Dev Biol. 2020 Dec 21;8:603688. doi: 10.3389/fcell.2020.603688. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 33409279 Free PMC article.
-
Arginyl-tRNA-protein transferase 1 (ATE1) promotes melanoma cell growth and migration.FEBS Lett. 2022 Jun;596(11):1468-1480. doi: 10.1002/1873-3468.14376. Epub 2022 May 20. FEBS Lett. 2022. PMID: 35561126 Free PMC article.
-
Availability of Arg, but Not tRNA, Is a Rate-Limiting Factor for Intracellular Arginylation.Int J Mol Sci. 2021 Dec 28;23(1):314. doi: 10.3390/ijms23010314. Int J Mol Sci. 2021. PMID: 35008737 Free PMC article.
-
Assaying Intracellular Arginylation Activity Using a Fluorescent Reporter.Methods Mol Biol. 2023;2620:81-85. doi: 10.1007/978-1-0716-2942-0_10. Methods Mol Biol. 2023. PMID: 37010751
References
-
- Kaji H, Novelli GD, Kaji A. A soluble amino acid-incorporating system from rat liver. Biochim Biophys Acta 1963; 76: 474–477. - PubMed
-
- Kwon YT, Kashina AS, Davydov IV, Hu RG, An JY, Seo JW et al. An essential role of N-terminal arginylation in cardiovascular development. Science 2002; 297: 96–99. - PubMed
-
- Balzi E, Choder M, Chen WN, Varshavsky A, Goffeau A. Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1 of Saccharomyces cerevisiae. J Biol Chem 1990; 265: 7464–7471. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
