

Identifying Significant Features in Cancer Methylation Data Using Gene Pathway Segmentation
- PMID: 27688706
- PMCID: PMC5030825
- DOI: 10.4137/CIN.S39859
Identifying Significant Features in Cancer Methylation Data Using Gene Pathway Segmentation
Abstract
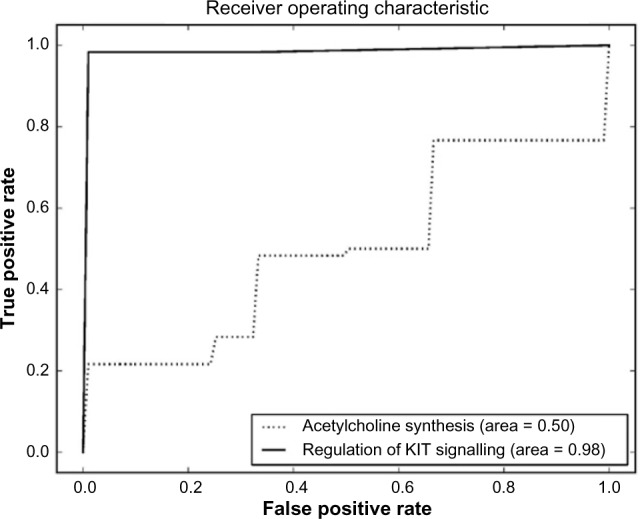
In order to provide the most effective therapy for cancer, it is important to be able to diagnose whether a patient's cancer will respond to a proposed treatment. Methylation profiling could contain information from which such predictions could be made. Currently, hypothesis testing is used to determine whether possible biomarkers for cancer progression produce statistically significant results. However, this approach requires the identification of individual genes, or sets of genes, as candidate hypotheses, and with the increasing size of modern microarrays, this task is becoming progressively harder. Exhaustive testing of small sets of genes is computationally infeasible, and so hypothesis generation depends either on the use of established biological knowledge or on heuristic methods. As an alternative machine learning, methods can be used to identify groups of genes that are acting together within sets of cancer data and associate their behaviors with cancer progression. These methods have the advantage of being multivariate and unbiased but unfortunately also rapidly become computationally infeasible as the number of gene probes and datasets increases. To address this problem, we have investigated a way of utilizing prior knowledge to segment microarray datasets in such a way that machine learning can be used to identify candidate sets of genes for hypothesis testing. A methylation dataset is divided into subsets, where each subset contains only the probes that relate to a known gene pathway. Each of these pathway subsets is used independently for classification. The classification method is AdaBoost with decision trees as weak classifiers. Since each pathway subset contains a relatively small number of gene probes, it is possible to train and test its classification accuracy quickly and determine whether it has valuable diagnostic information. Finally, genes from successful pathway subsets can be combined to create a classifier of high accuracy.
Keywords: cancer progression; machine learning; methylation profiling.
Figures

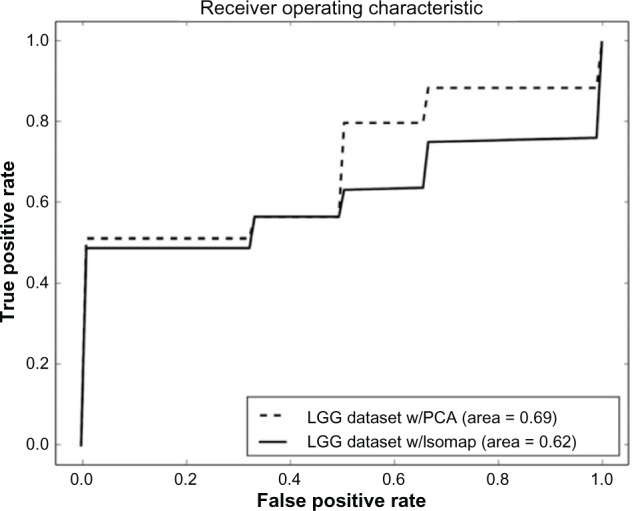
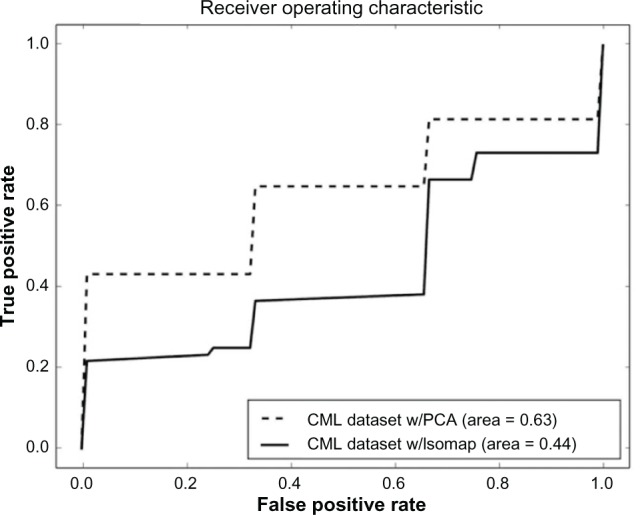
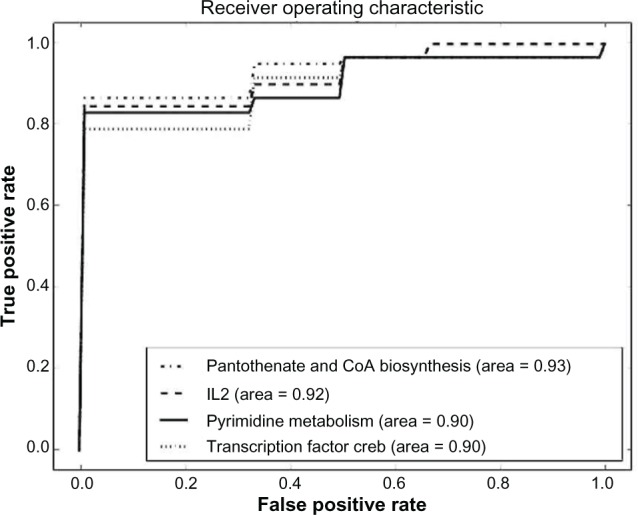
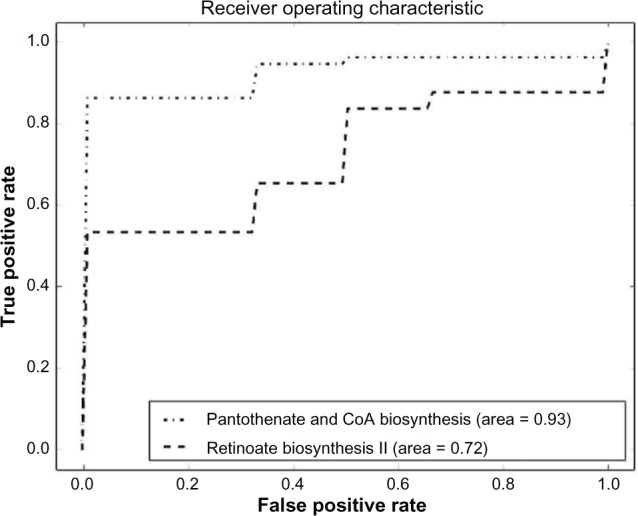
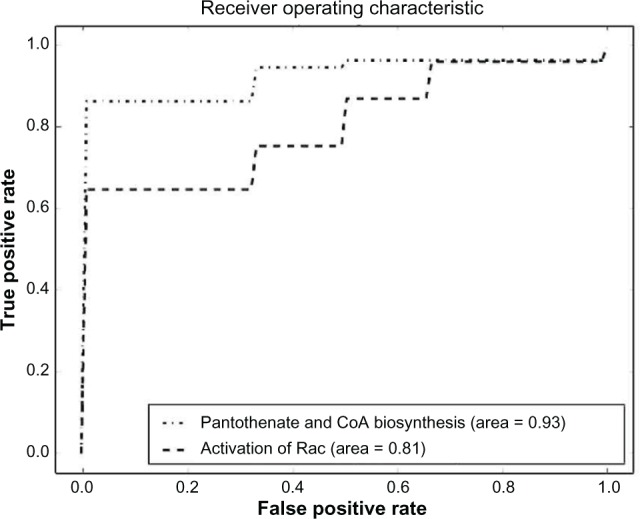

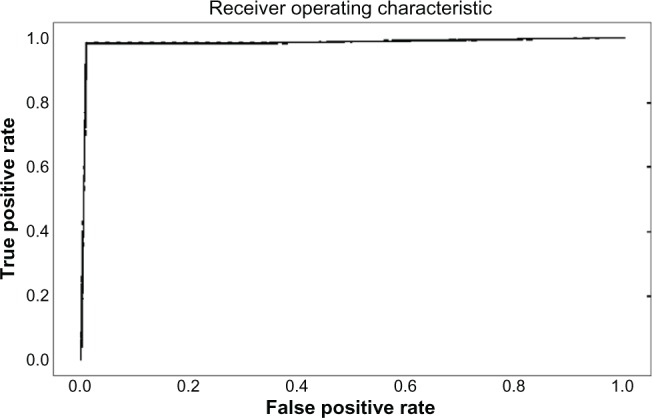
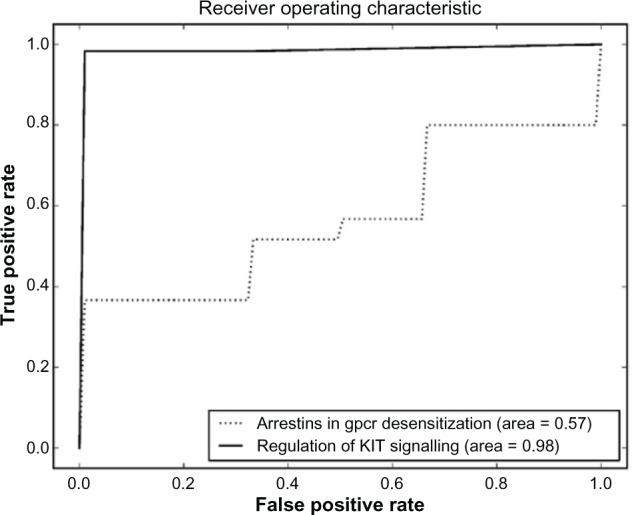
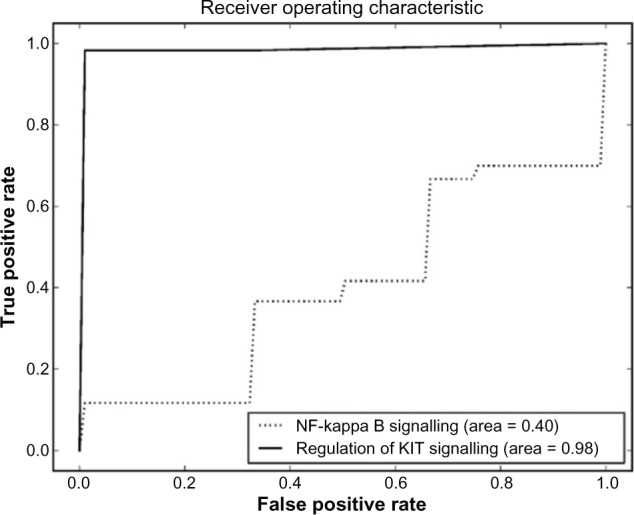
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
