

Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism
- PMID: 27688759
- PMCID: PMC5081600
- DOI: 10.1073/pnas.1607496113
Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism
Abstract
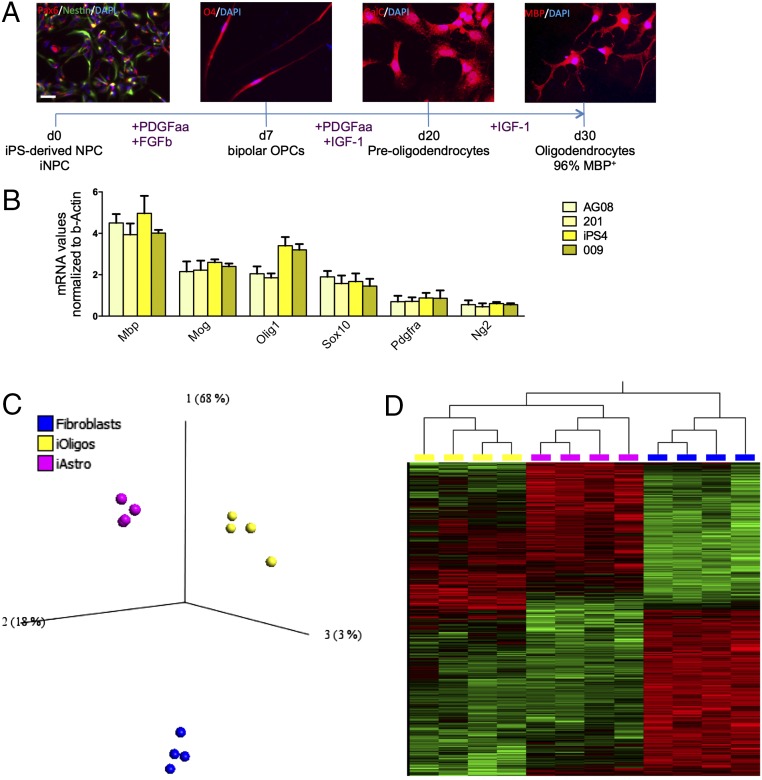
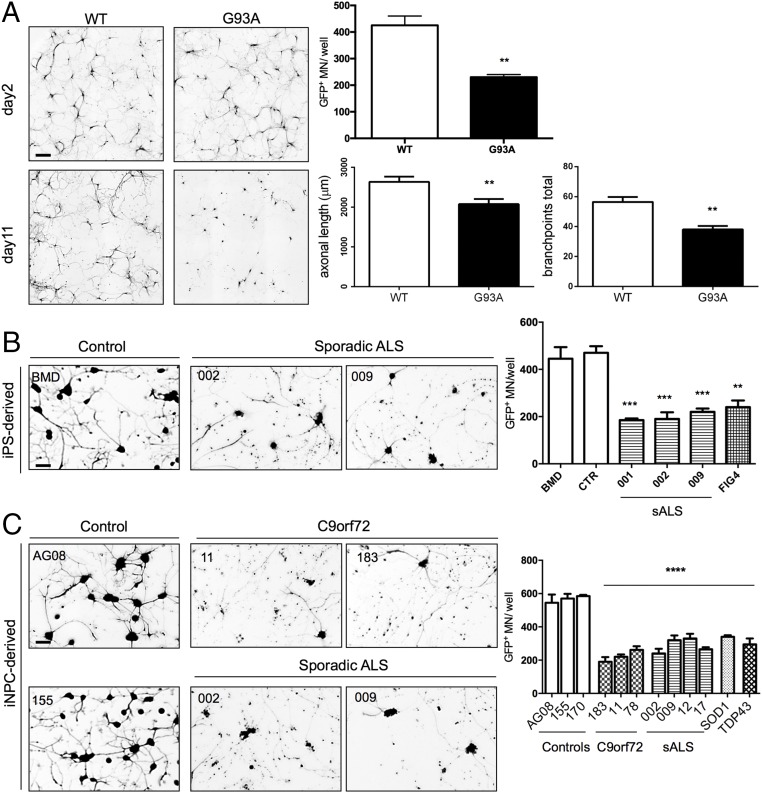
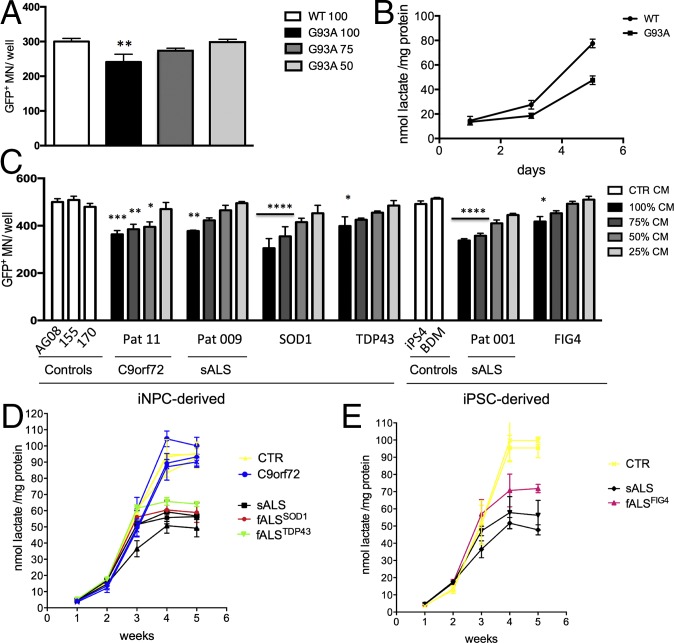
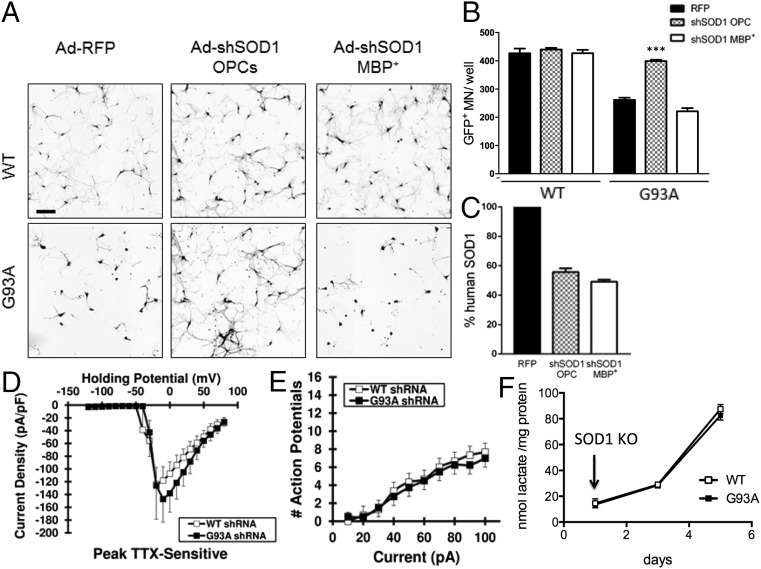
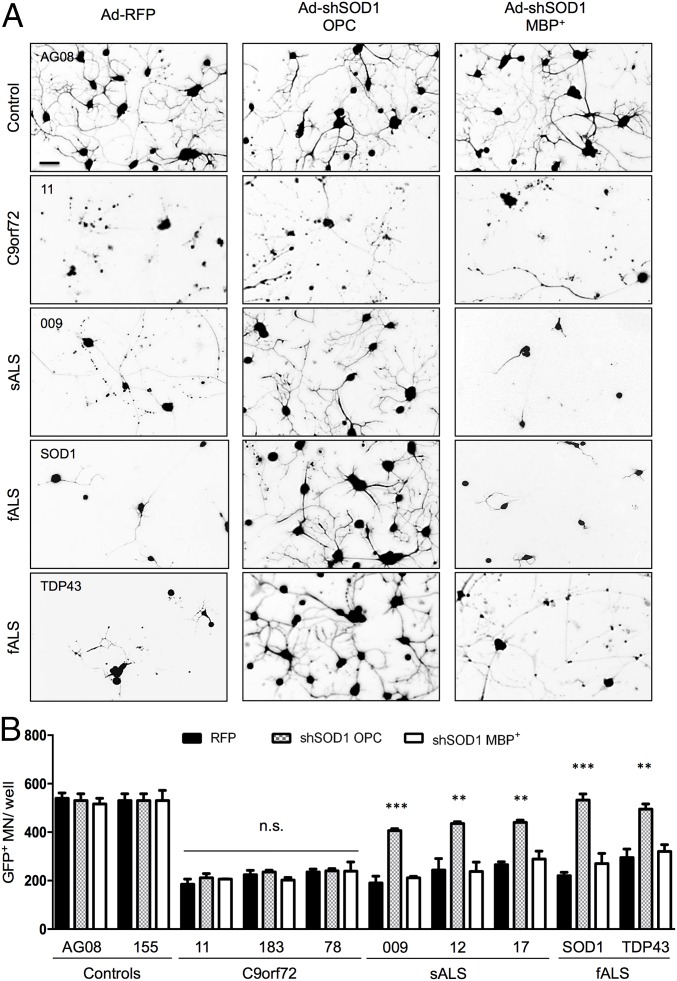
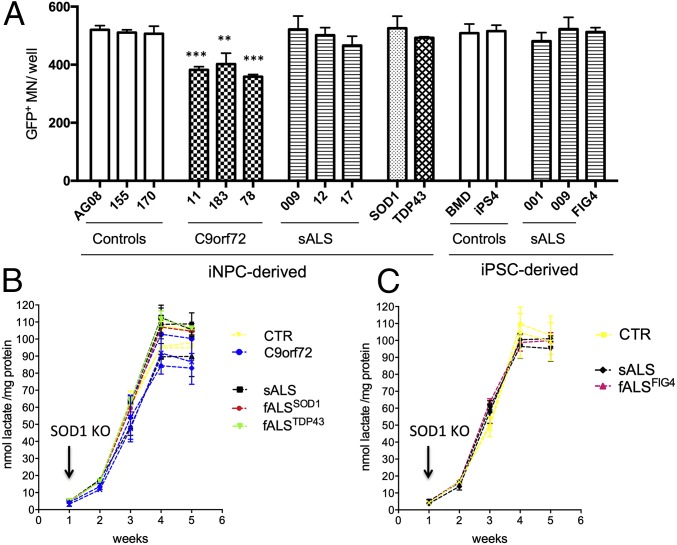
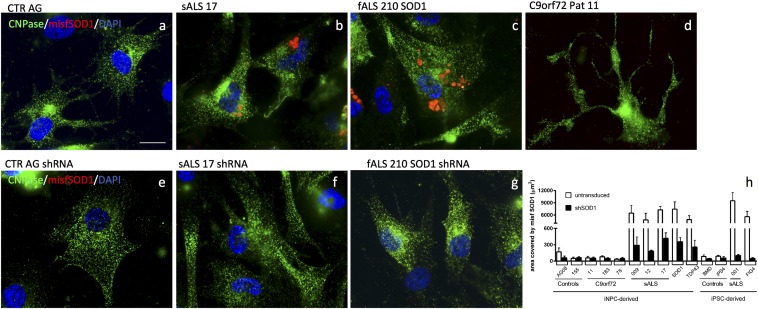
Oligodendrocytes have recently been implicated in the pathophysiology of amyotrophic lateral sclerosis (ALS). Here we show that, in vitro, mutant superoxide dismutase 1 (SOD1) mouse oligodendrocytes induce WT motor neuron (MN) hyperexcitability and death. Moreover, we efficiently derived human oligodendrocytes from a large number of controls and patients with sporadic and familial ALS, using two different reprogramming methods. All ALS oligodendrocyte lines induced MN death through conditioned medium (CM) and in coculture. CM-mediated MN death was associated with decreased lactate production and release, whereas toxicity in coculture was lactate-independent, demonstrating that MN survival is mediated not only by soluble factors. Remarkably, human SOD1 shRNA treatment resulted in MN rescue in both mouse and human cultures when knockdown was achieved in progenitor cells, whereas it was ineffective in differentiated oligodendrocytes. In fact, early SOD1 knockdown rescued lactate impairment and cell toxicity in all lines tested, with the exclusion of samples carrying chromosome 9 ORF 72 (C9orf72) repeat expansions. These did not respond to SOD1 knockdown nor did they show lactate release impairment. Our data indicate that SOD1 is directly or indirectly involved in ALS oligodendrocyte pathology and suggest that in this cell type, some damage might be irreversible. In addition, we demonstrate that patients with C9ORF72 represent an independent patient group that might not respond to the same treatment.
Keywords: C9orf72; SOD1; amyotrophic lateral sclerosis; lactate; oligodendrocytes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;364(6435):362. - PubMed
-
- Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. - PubMed
-
- Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
