

De Novo Truncating Variants in ASXL2 Are Associated with a Unique and Recognizable Clinical Phenotype
- PMID: 27693232
- PMCID: PMC5065681
- DOI: 10.1016/j.ajhg.2016.08.017
De Novo Truncating Variants in ASXL2 Are Associated with a Unique and Recognizable Clinical Phenotype
Erratum in
-
De Novo Truncating Variants in ASXL2 Are Associated with a Unique and Recognizable Clinical Phenotype.Am J Hum Genet. 2017 Jan 5;100(1):179. doi: 10.1016/j.ajhg.2016.12.004. Am J Hum Genet. 2017. PMID: 28061364 Free PMC article. No abstract available.
Abstract
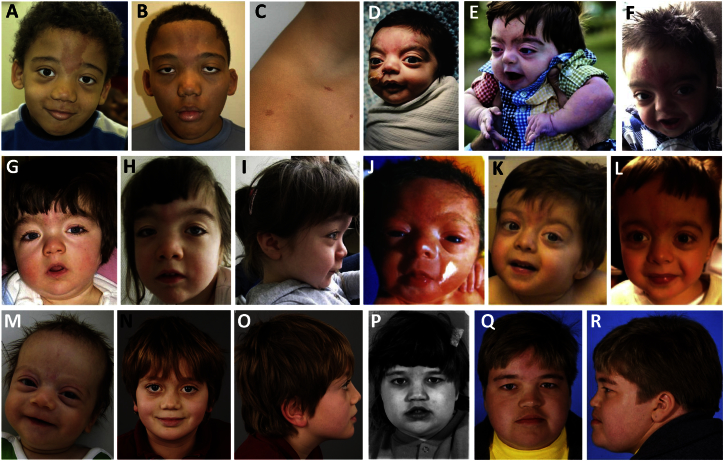
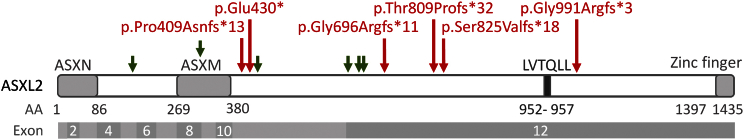
The ASXL genes (ASXL1, ASXL2, and ASXL3) participate in body patterning during embryogenesis and encode proteins involved in epigenetic regulation and assembly of transcription factors to specific genomic loci. Germline de novo truncating variants in ASXL1 and ASXL3 have been respectively implicated in causing Bohring-Opitz and Bainbridge-Ropers syndromes, which result in overlapping features of severe intellectual disability and dysmorphic features. ASXL2 has not yet been associated with a human Mendelian disorder. In this study, we performed whole-exome sequencing in six unrelated probands with developmental delay, macrocephaly, and dysmorphic features. All six had de novo truncating variants in ASXL2. A careful review enabled the recognition of a specific phenotype consisting of macrocephaly, prominent eyes, arched eyebrows, hypertelorism, a glabellar nevus flammeus, neonatal feeding difficulties, hypotonia, and developmental disabilities. Although overlapping features with Bohring-Opitz and Bainbridge-Ropers syndromes exist, features that distinguish the ASXL2-associated condition from ASXL1- and ASXL3-related disorders are macrocephaly, absence of growth retardation, and more variability in the degree of intellectual disabilities. We were also able to demonstrate with mRNA studies that these variants are likely to exert a dominant-negative effect, given that both alleles are expressed in blood and the mutated ASXL2 transcripts escape nonsense-mediated decay. In conclusion, de novo truncating variants in ASXL2 underlie a neurodevelopmental syndrome with a clinically recognizable phenotype. This report expands the germline disorders that are linked to the ASXL genes.
Keywords: ASXL2; developmental delay; glabellar nevus flammeus; intellectual disability; macrocephaly; whole-exome sequencing.
Copyright © 2016. Published by Elsevier Inc.
Figures
References
-
- Fisher C.L., Berger J., Randazzo F., Brock H.W. A human homolog of Additional sex combs, ADDITIONAL SEX COMBS-LIKE 1, maps to chromosome 20q11. Gene. 2003;306:115–126. - PubMed
-
- Katoh M. Functional proteomics of the epigenetic regulators ASXL1, ASXL2 and ASXL3: a convergence of proteomics and epigenetics for translational medicine. Expert Rev. Proteomics. 2015;12:317–328. - PubMed
-
- Katoh M., Katoh M. Identification and characterization of ASXL3 gene in silico. Int. J. Oncol. 2004;24:1617–1622. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
