

A systematic evaluation of normalization methods in quantitative label-free proteomics
- PMID: 27694351
- PMCID: PMC5862339
- DOI: 10.1093/bib/bbw095
A systematic evaluation of normalization methods in quantitative label-free proteomics
Abstract
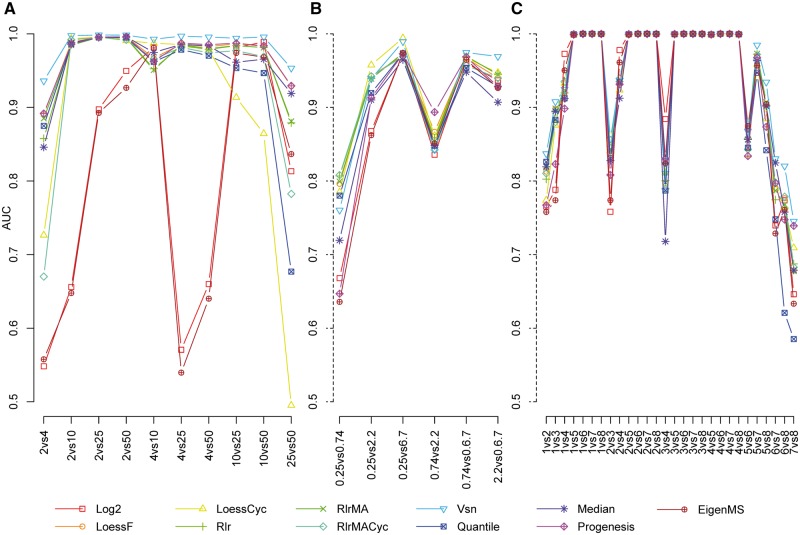
To date, mass spectrometry (MS) data remain inherently biased as a result of reasons ranging from sample handling to differences caused by the instrumentation. Normalization is the process that aims to account for the bias and make samples more comparable. The selection of a proper normalization method is a pivotal task for the reliability of the downstream analysis and results. Many normalization methods commonly used in proteomics have been adapted from the DNA microarray techniques. Previous studies comparing normalization methods in proteomics have focused mainly on intragroup variation. In this study, several popular and widely used normalization methods representing different strategies in normalization are evaluated using three spike-in and one experimental mouse label-free proteomic data sets. The normalization methods are evaluated in terms of their ability to reduce variation between technical replicates, their effect on differential expression analysis and their effect on the estimation of logarithmic fold changes. Additionally, we examined whether normalizing the whole data globally or in segments for the differential expression analysis has an effect on the performance of the normalization methods. We found that variance stabilization normalization (Vsn) reduced variation the most between technical replicates in all examined data sets. Vsn also performed consistently well in the differential expression analysis. Linear regression normalization and local regression normalization performed also systematically well. Finally, we discuss the choice of a normalization method and some qualities of a suitable normalization method in the light of the results of our evaluation.
Keywords: bias; differential expression; intragroup variation; label free; logarithmic fold change; mass spectrometry; normalization; proteomics; quantitation; reproducibility.
© The Author 2016. Published by Oxford University Press.
Figures




References
-
- Megger DA, Bracht T, Meyer HE, et al. Label-free quantification in clinical proteomics. Biochim Biophys Acta 2013;1834:1581–90. - PubMed
-
- Meissner F, Mann M.. Quantitative shotgun proteomics: considerations for a high-quality workflow in immunology. Nat Immunol 2014;15:112–7. - PubMed
-
- Bolstad BM, Irizarry RA, Åstrand M, et al. A comparison of normalization methods for high density Oligonucleotide array data based on variance and bias. Bioinformatics 2003;19:185–93. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
