

Identification and outcomes of clinical phenotypes in amyotrophic lateral sclerosis/motor neuron disease: Australian National Motor Neuron Disease observational cohort
- PMID: 27694488
- PMCID: PMC5051496
- DOI: 10.1136/bmjopen-2016-012054
Identification and outcomes of clinical phenotypes in amyotrophic lateral sclerosis/motor neuron disease: Australian National Motor Neuron Disease observational cohort
Abstract
Objective: To capture the clinical patterns, timing of key milestones and survival of patients presenting with amyotrophic lateral sclerosis/motor neuron disease (ALS/MND) within Australia.
Methods: Data were prospectively collected and were timed to normal clinical assessments. An initial registration clinical report form (CRF) and subsequent ongoing assessment CRFs were submitted with a completion CRF at the time of death.
Design: Prospective observational cohort study.
Participants: 1834 patients with a diagnosis of ALS/MND were registered and followed in ALS/MND clinics between 2005 and 2015.
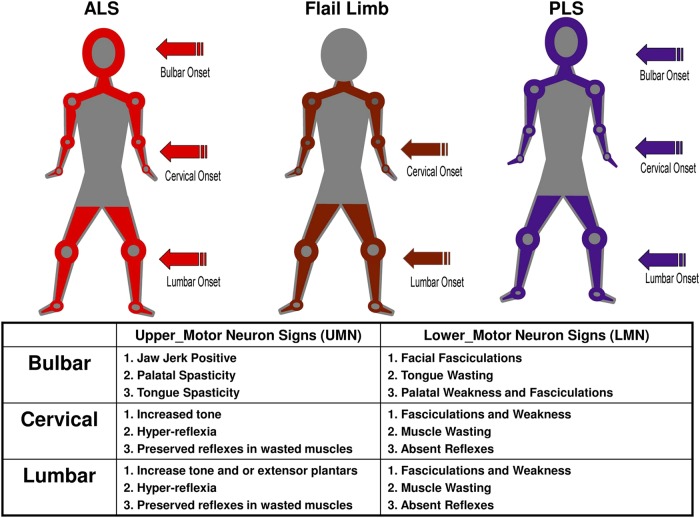
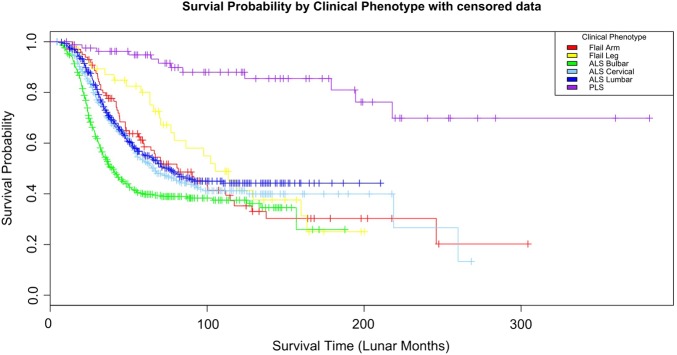
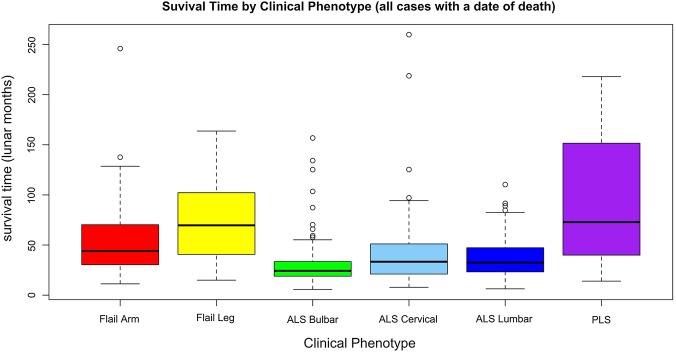
Results: 5 major clinical phenotypes were determined and included ALS bulbar onset, ALS cervical onset and ALS lumbar onset, flail arm and leg and primary lateral sclerosis (PLS). Of the 1834 registered patients, 1677 (90%) could be allocated a clinical phenotype. ALS bulbar onset had a significantly lower length of survival when compared with all other clinical phenotypes (p<0.004). There were delays in the median time to diagnosis of up to 12 months for the ALS phenotypes, 18 months for the flail limb phenotypes and 19 months for PLS. Riluzole treatment was started in 78-85% of cases. The median delays in initiating riluzole therapy, from symptom onset, varied from 10 to 12 months in the ALS phenotypes and 15-18 months in the flail limb phenotypes. Percutaneous endoscopic gastrostomy was implemented in 8-36% of ALS phenotypes and 2-9% of the flail phenotypes. Non-invasive ventilation was started in 16-22% of ALS phenotypes and 21-29% of flail phenotypes.
Conclusions: The establishment of a cohort registry for ALS/MND is able to determine clinical phenotypes, survival and monitor time to key milestones in disease progression. It is intended to expand the cohort to a more population-based registry using opt-out methodology and facilitate data linkage to other national registries.
Keywords: EPIDEMIOLOGY.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/.
Figures
References
-
- Brooks BR. El Escorial World Federation of Neurology criteria for the daignosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Disease/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial ‘Clinical Limits of amyotrophic lateral sclerosis’ workshop contributors. J Neurol Sci 1994;124(Suppl):96–107. - PubMed
-
- World Federation of Neurology Research Group on Neuromuscular Diseases Subcommittee on Motor Neuron Disease. Airlie House Guidelines. Therapeutic trials in amyotrophic lateral sclerosis. Airlie House ‘Therapeutic trials in ALS’ Workshop Contributors. J Neurol Sci 1995;129:1–10. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous