

Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis
- PMID: 27694913
- PMCID: PMC5343289
- DOI: 10.1038/nri.2016.100
Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis
Abstract
Autophagy has broad functions in immunity, ranging from cell-autonomous defence to coordination of complex multicellular immune responses. The successful resolution of infection and avoidance of autoimmunity necessitates efficient and timely communication between autophagy and pathways that sense the immune environment. The recent literature indicates that a variety of immune mediators induce or repress autophagy. It is also becoming increasingly clear that immune signalling cascades are subject to regulation by autophagy, and that a return to homeostasis following a robust immune response is critically dependent on this pathway. Importantly, examples of non-canonical forms of autophagy in mediating immunity are pervasive. In this article, the progress in elucidating mechanisms of crosstalk between autophagy and inflammatory signalling cascades is reviewed. Improved mechanistic understanding of the autophagy machinery offers hope for treating infectious and inflammatory diseases.
Conflict of interest statement
The author declares no competing financial interests.
Figures
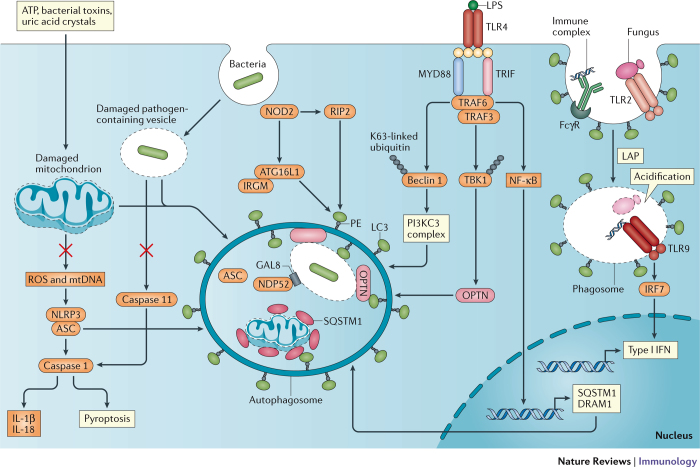
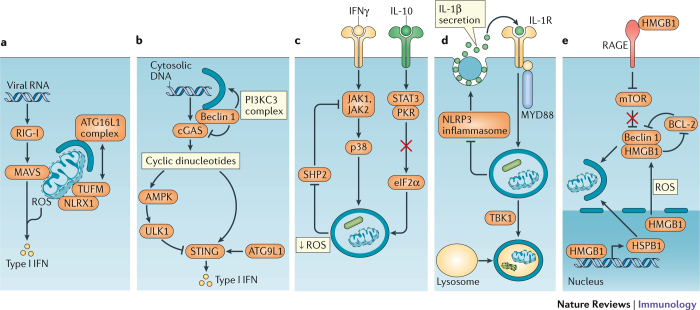
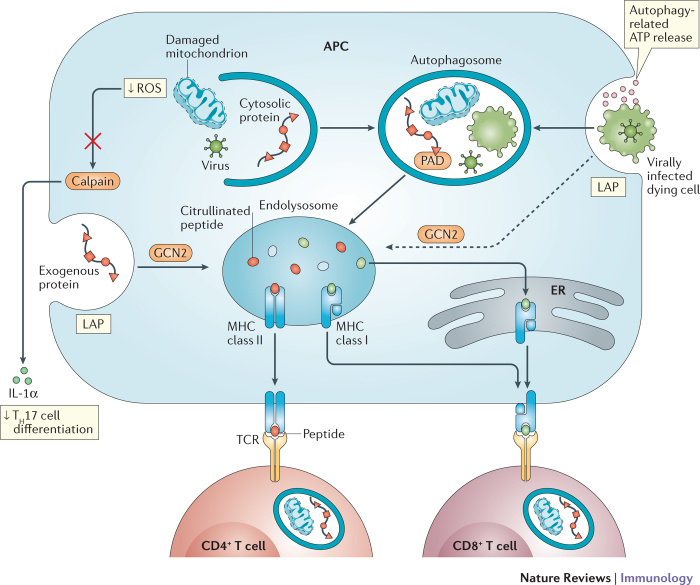
References
-
- Hamasaki M, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
