

Putative Receptor Binding Domain of Bat-Derived Coronavirus HKU9 Spike Protein: Evolution of Betacoronavirus Receptor Binding Motifs
- PMID: 27696819
- PMCID: PMC7075523
- DOI: 10.1021/acs.biochem.6b00790
Putative Receptor Binding Domain of Bat-Derived Coronavirus HKU9 Spike Protein: Evolution of Betacoronavirus Receptor Binding Motifs
Abstract
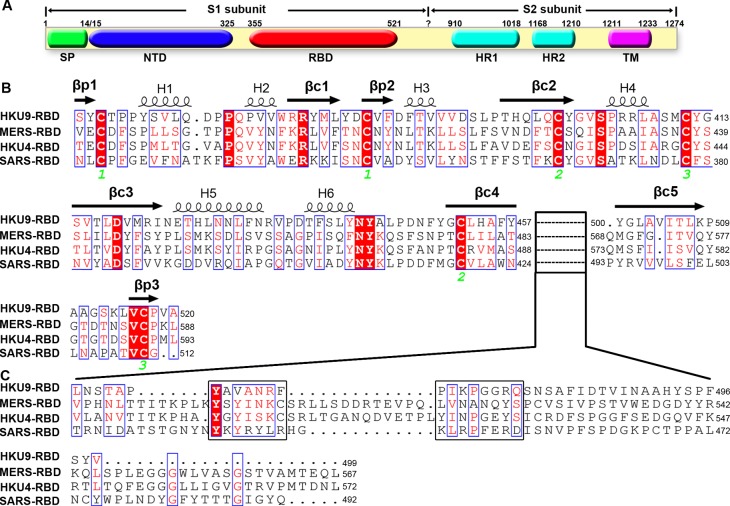
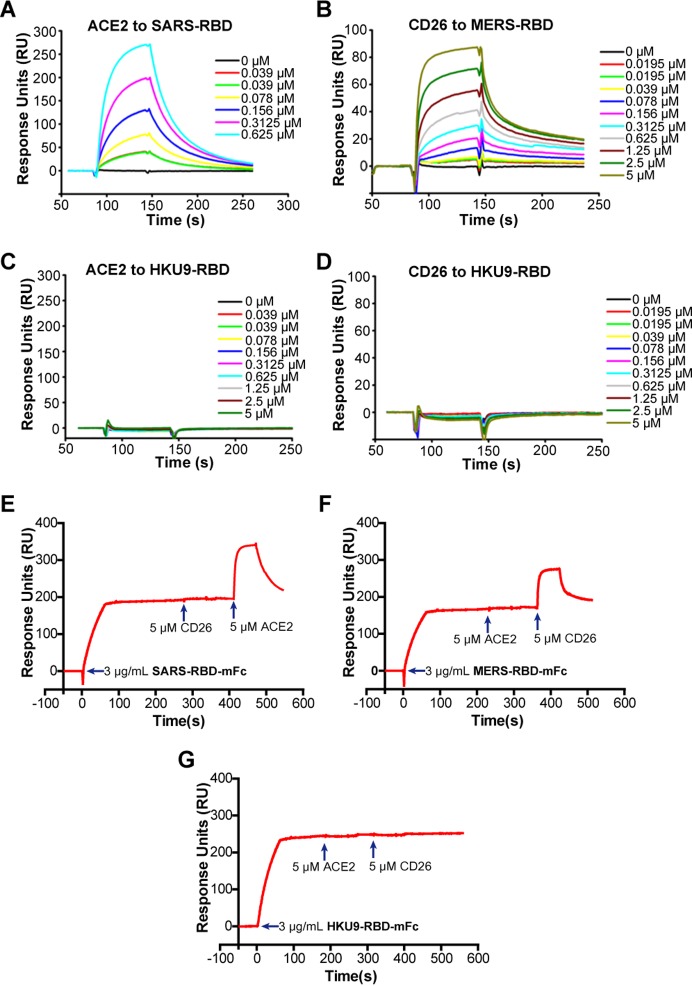
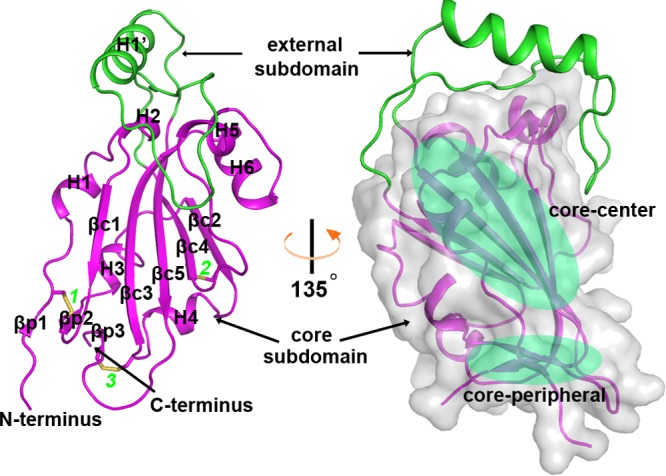
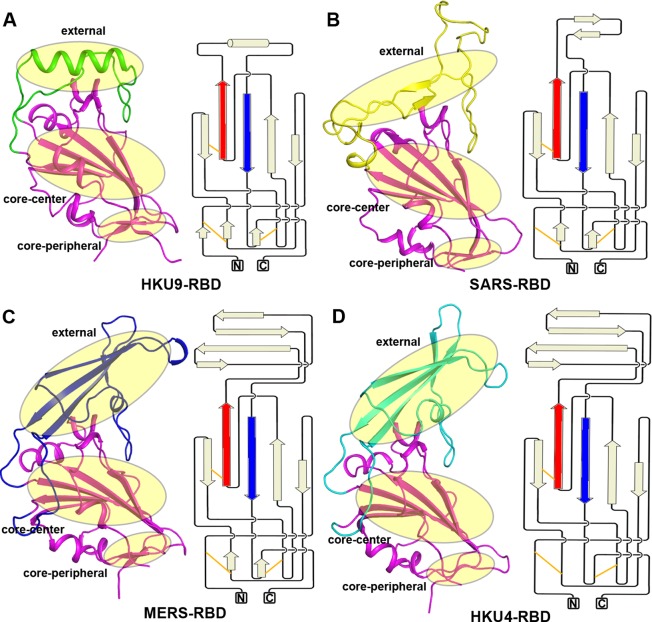
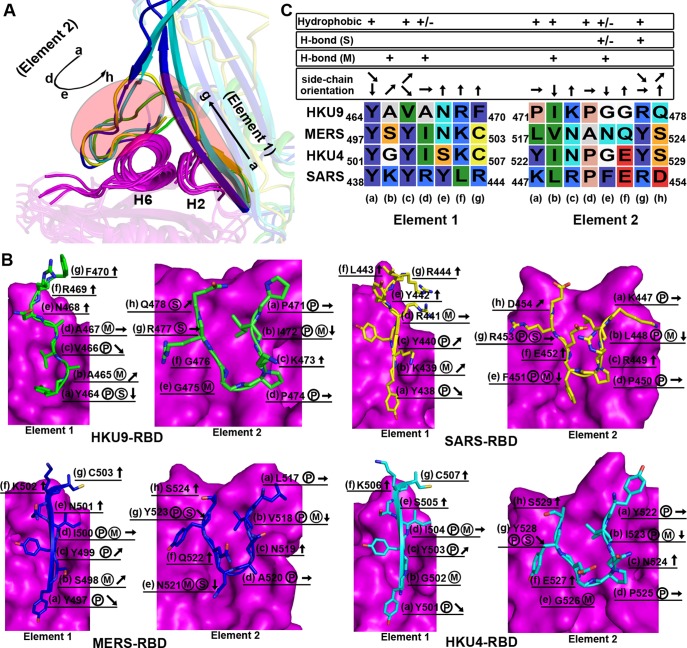
The suggested bat origin for Middle East respiratory syndrome coronavirus (MERS-CoV) has revitalized the studies of other bat-derived coronaviruses with respect to interspecies transmission potential. Bat coronavirus (BatCoV) HKU9 is an important betacoronavirus (betaCoV) that is phylogenetically affiliated with the same genus as MERS-CoV. The bat surveillance data indicated that BatCoV HKU9 has been widely spreading and circulating in bats. This highlights the necessity of characterizing the virus for its potential to cross species barriers. The receptor binding domain (RBD) of the coronavirus spike (S) protein recognizes host receptors to mediate virus entry and is therefore a key factor determining the viral tropism and transmission capacity. In this study, the putative S RBD of BatCoV HKU9 (HKU9-RBD), which is homologous to other betaCoV RBDs that have been structurally and functionally defined, was characterized via a series of biophysical and crystallographic methods. By using surface plasmon resonance, we demonstrated that HKU9-RBD binds to neither SARS-CoV receptor ACE2 nor MERS-CoV receptor CD26. We further determined the atomic structure of HKU9-RBD, which as expected is composed of a core and an external subdomain. The core subdomain fold resembles those of other betaCoV RBDs, whereas the external subdomain is structurally unique with a single helix, explaining the inability of HKU9-RBD to react with either ACE2 or CD26. Via comparison of the available RBD structures, we further proposed a homologous intersubdomain binding mode in betaCoV RBDs that anchors the external subdomain to the core subdomain. The revealed RBD features would shed light on the evolution route of betaCoV.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
Detection and full genome characterization of two beta CoV viruses related to Middle East respiratory syndrome from bats in Italy.Virol J. 2017 Dec 19;14(1):239. doi: 10.1186/s12985-017-0907-1. Virol J. 2017. PMID: 29258555 Free PMC article.
-
Receptor Usage of a Novel Bat Lineage C Betacoronavirus Reveals Evolution of Middle East Respiratory Syndrome-Related Coronavirus Spike Proteins for Human Dipeptidyl Peptidase 4 Binding.J Infect Dis. 2018 Jun 20;218(2):197-207. doi: 10.1093/infdis/jiy018. J Infect Dis. 2018. PMID: 29346682 Free PMC article.
-
Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells.J Biol Chem. 2018 Jul 27;293(30):11709-11726. doi: 10.1074/jbc.RA118.001897. Epub 2018 Jun 10. J Biol Chem. 2018. PMID: 29887526 Free PMC article.
-
Advances in MERS-CoV Vaccines and Therapeutics Based on the Receptor-Binding Domain.Viruses. 2019 Jan 14;11(1):60. doi: 10.3390/v11010060. Viruses. 2019. PMID: 30646569 Free PMC article. Review.
-
A structural view of coronavirus-receptor interactions.Virus Res. 2014 Dec 19;194:3-15. doi: 10.1016/j.virusres.2014.10.005. Epub 2014 Oct 14. Virus Res. 2014. PMID: 25451063 Free PMC article. Review.
Cited by
-
Coronaviruses spike glycoprotein endodomains: The sequence and structure-based comprehensive study.Protein Sci. 2023 Nov;32(11):e4804. doi: 10.1002/pro.4804. Protein Sci. 2023. PMID: 37833239 Free PMC article.
-
Cryo-EM structures of HKU2 and SADS-CoV spike glycoproteins provide insights into coronavirus evolution.Nat Commun. 2020 Jun 17;11(1):3070. doi: 10.1038/s41467-020-16876-4. Nat Commun. 2020. PMID: 32555182 Free PMC article.
-
In-silico investigation of systematic missense mutations of middle east respiratory coronavirus spike protein.Front Mol Biosci. 2022 Sep 14;9:933553. doi: 10.3389/fmolb.2022.933553. eCollection 2022. Front Mol Biosci. 2022. PMID: 36188214 Free PMC article.
-
The Physical Basis for pH Sensitivity in Biomolecular Structure and Function, With Application to the Spike Protein of SARS-CoV-2.Front Mol Biosci. 2022 Feb 18;9:834011. doi: 10.3389/fmolb.2022.834011. eCollection 2022. Front Mol Biosci. 2022. PMID: 35252354 Free PMC article. Review.
-
Unveiling hidden structural patterns in the SARS-CoV-2 genome: Computational insights and comparative analysis.PLoS One. 2024 Apr 4;19(4):e0298164. doi: 10.1371/journal.pone.0298164. eCollection 2024. PLoS One. 2024. PMID: 38574063 Free PMC article.
References
-
- Fields B. N., Knipe D. M., and Howley P. M. (2007) Fields virology, 5th ed., Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia.
-
- International Committee on Taxonomy of Viruses and King A. M. Q. (2012) Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses, Academic Press, London.
-
- Schalk A. F.; Hawn M. C. (1931) An apparently new respiratory disease in baby chicks. J. Am. Vet. Med. Assoc. 78, 413–422.
-
- Woo P. C.; Lau S. K.; Lam C. S.; Lai K. K.; Huang Y.; Lee P.; Luk G. S.; Dyrting K. C.; Chan K. H.; Yuen K. Y. (2009) Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J. Virol. 83, 908–917. 10.1128/JVI.01977-08. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
