

Cell Signaling and Stress Responses
- PMID: 27698029
- PMCID: PMC5046695
- DOI: 10.1101/cshperspect.a006072
Cell Signaling and Stress Responses
Abstract
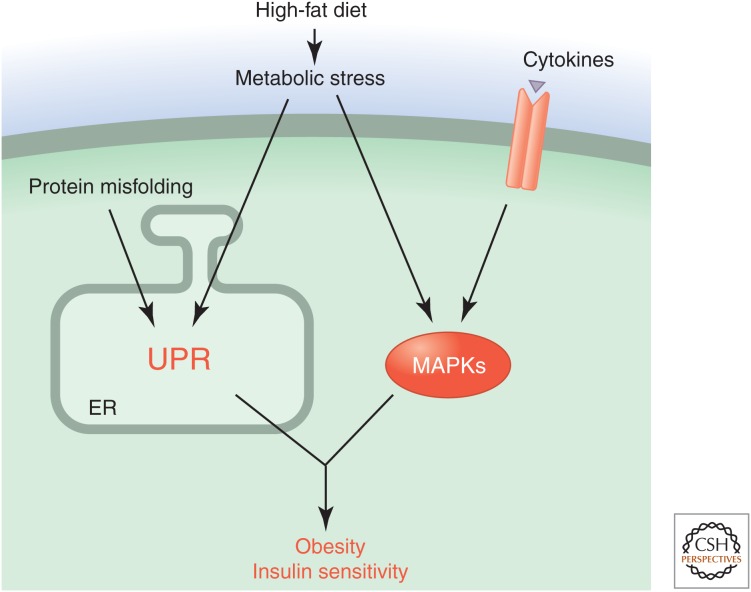
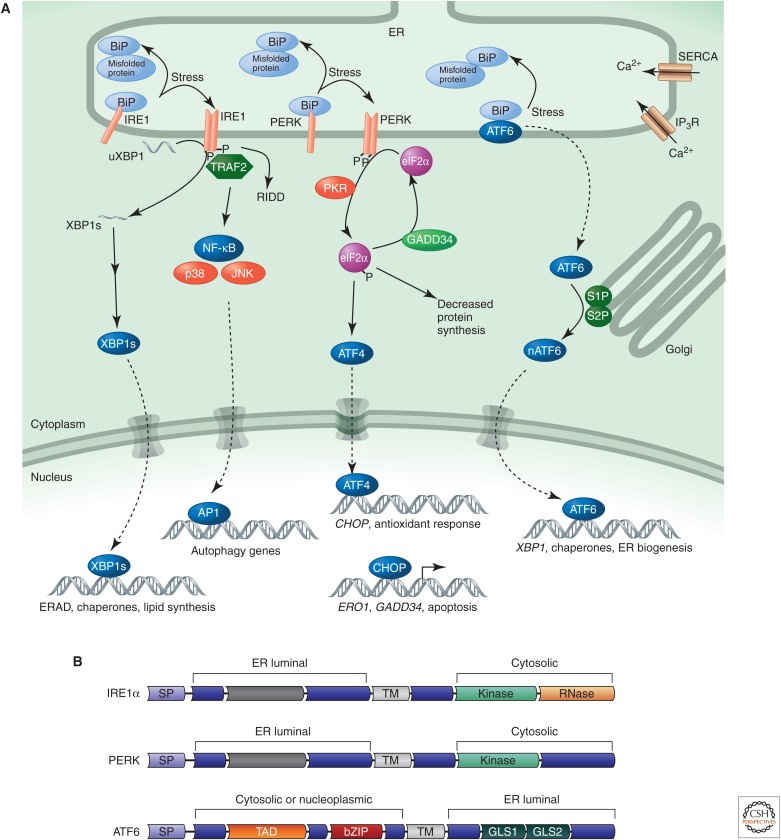
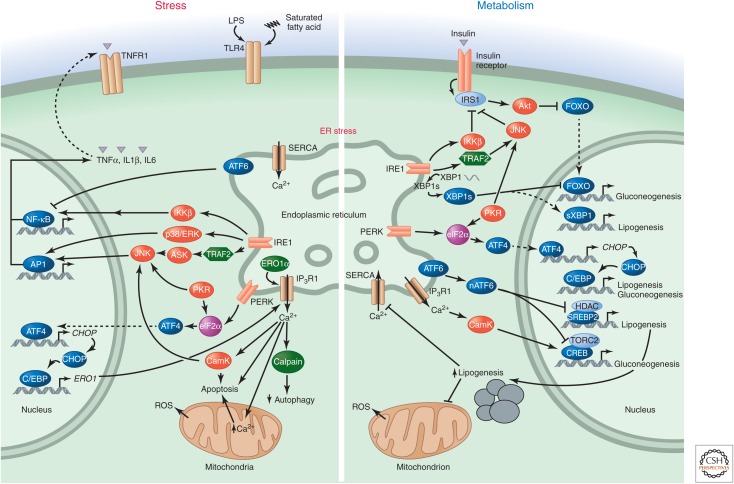
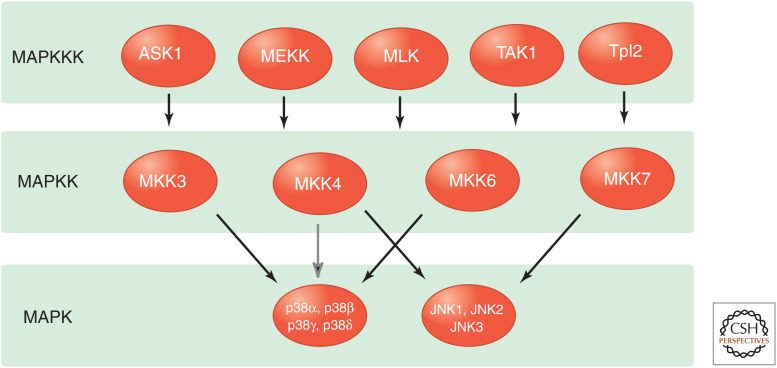
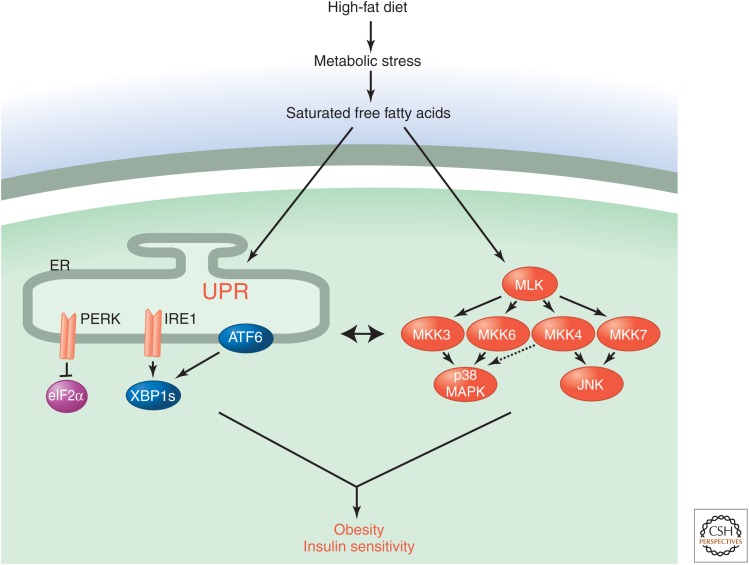
Stress-signaling pathways are evolutionarily conserved and play an important role in the maintenance of homeostasis. These pathways are also critical for adaptation to new cellular environments. The endoplasmic reticulum (ER) unfolded protein response (UPR) is activated by biosynthetic stress and leads to a compensatory increase in ER function. The JNK and p38 MAPK signaling pathways control adaptive responses to intracellular and extracellular stresses, including environmental changes such as UV light, heat, and hyperosmotic conditions, and exposure to inflammatory cytokines. Metabolic stress caused by a high-fat diet represents an example of a stimulus that coordinately activates both the UPR and JNK/p38 signaling pathways. Chronic activation of these stress-response pathways ultimately causes metabolic changes associated with obesity and altered insulin sensitivity. Stress-signaling pathways, therefore, represent potential targets for therapeutic intervention in the metabolic stress response and other disease processes.
Copyright © 2016 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Abe MK, Kahle KT, Saelzler MP, Orth K, Dixon JE, Rosner MR. 2001. ERK7 is an autoactivated member of the MAPK family. J Biol Chem 276: 21272–21279. - PubMed
-
- Aguirre V, Uchida T, Yenush L, Davis R, White MF. 2000. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J Biol Chem 275: 9047–9054. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials