

A multicenter study benchmarks software tools for label-free proteome quantification
- PMID: 27701404
- PMCID: PMC5120688
- DOI: 10.1038/nbt.3685
A multicenter study benchmarks software tools for label-free proteome quantification
Abstract
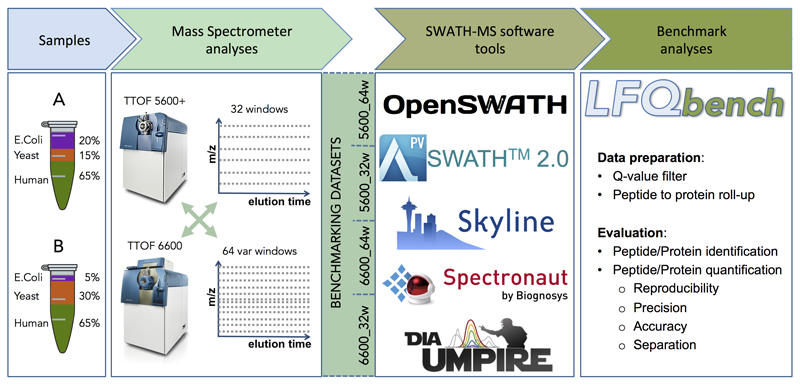
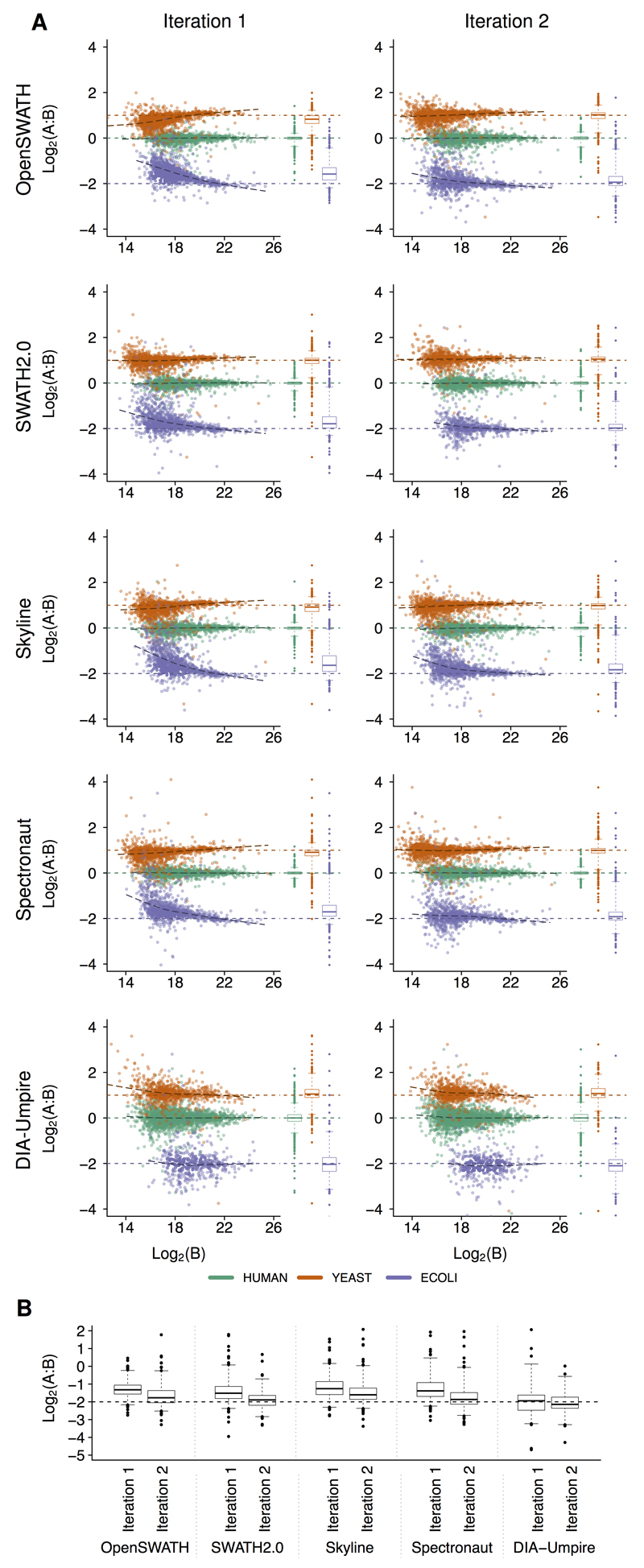
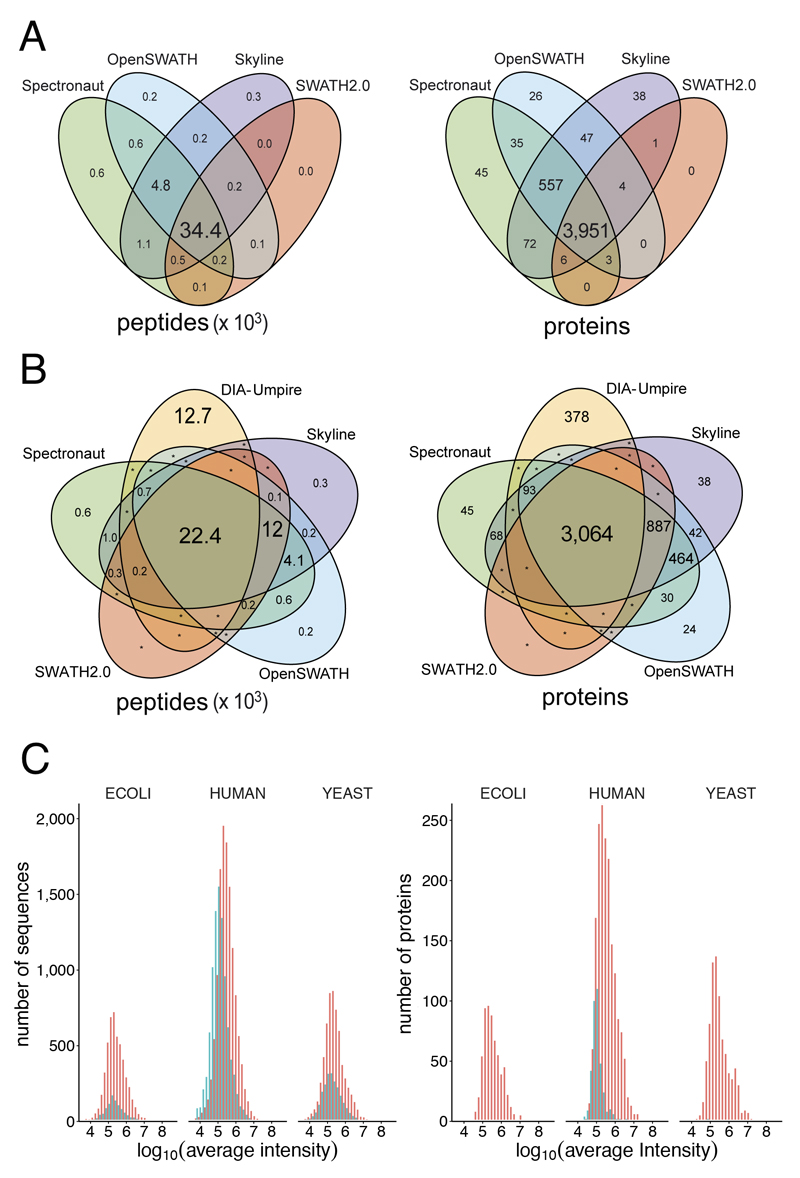
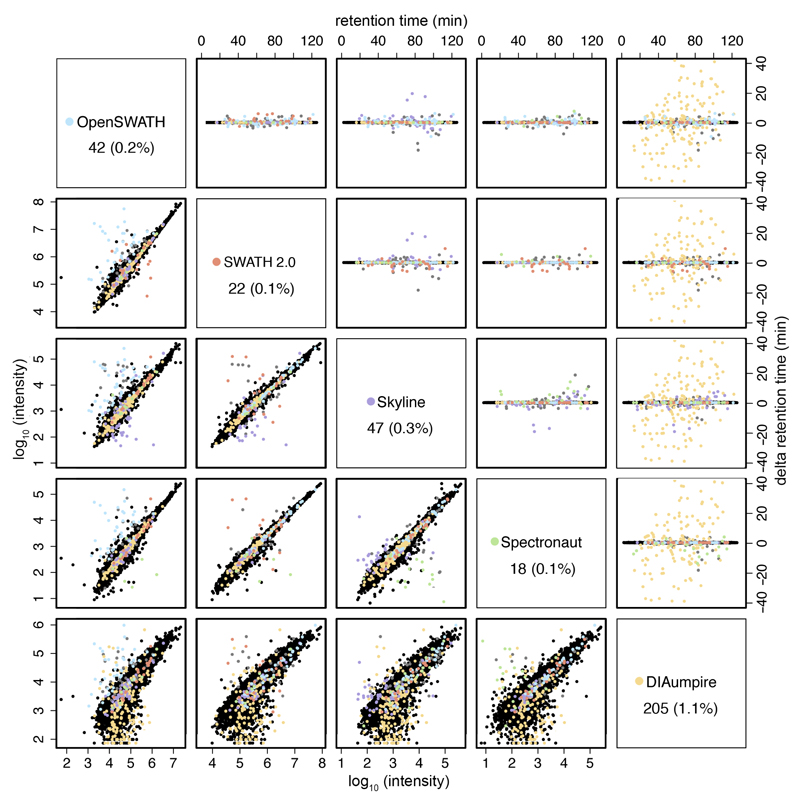
Consistent and accurate quantification of proteins by mass spectrometry (MS)-based proteomics depends on the performance of instruments, acquisition methods and data analysis software. In collaboration with the software developers, we evaluated OpenSWATH, SWATH 2.0, Skyline, Spectronaut and DIA-Umpire, five of the most widely used software methods for processing data from sequential window acquisition of all theoretical fragment-ion spectra (SWATH)-MS, which uses data-independent acquisition (DIA) for label-free protein quantification. We analyzed high-complexity test data sets from hybrid proteome samples of defined quantitative composition acquired on two different MS instruments using different SWATH isolation-window setups. For consistent evaluation, we developed LFQbench, an R package, to calculate metrics of precision and accuracy in label-free quantitative MS and report the identification performance, robustness and specificity of each software tool. Our reference data sets enabled developers to improve their software tools. After optimization, all tools provided highly convergent identification and reliable quantification performance, underscoring their robustness for label-free quantitative proteomics.
Conflict of interest statement
The authors declare the following competing financial interests: S.A.T. is employed by SCIEX, O.M.B. and L.R. are employed by Biognosys AG.
Figures
References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Mallick P, Kuster B. Proteomics: a pragmatic perspective. Nat Biotechnol. 2010;28:695–709. - PubMed
-
- Distler U, Kuharev J, Tenzer S. Biomedical applications of ion mobility-enhanced data-independent acquisition-based label-free quantitative proteomics. Expert Rev Proteomics. 2014;11:675–684. - PubMed
-
- Geromanos SJ, Hughes C, Ciavarini S, Vissers JPC, Langridge JI. Using ion purity scores for enhancing quantitative accuracy and precision in complex proteomics samples. Anal Bioanal Chem. 2012;404:1127–1139. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
