

Lafora disease
- PMID: 27702709
- PMCID: PMC5777303
- DOI: 10.1684/epd.2016.0842
Lafora disease
Abstract
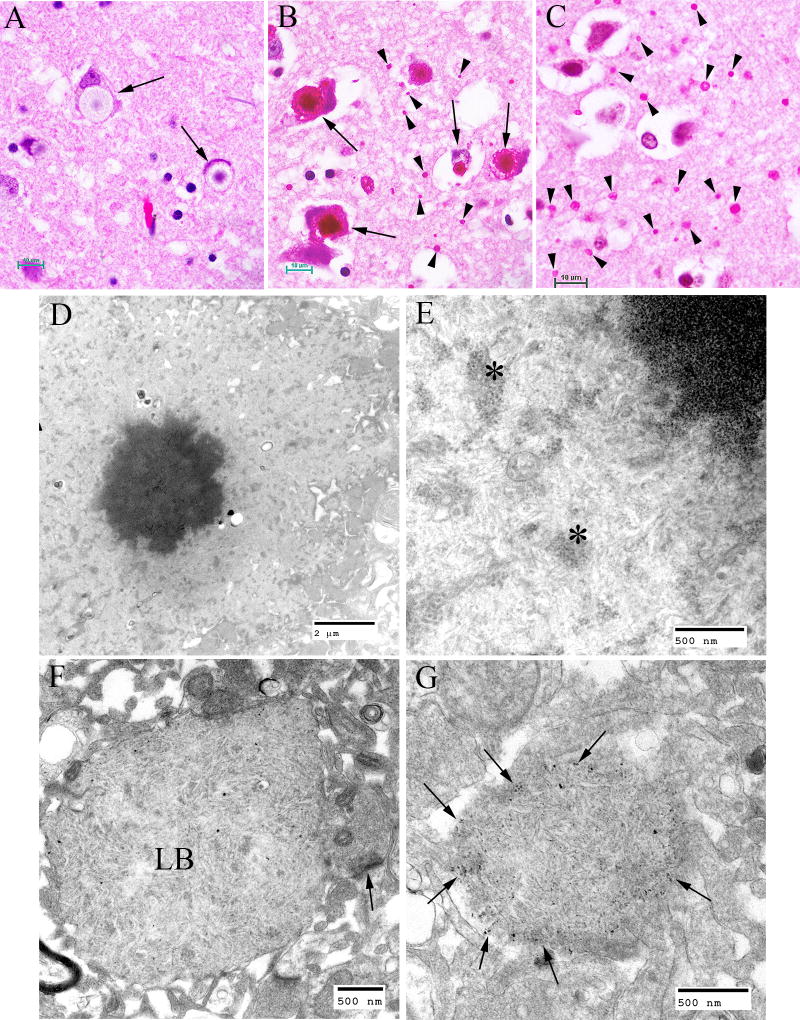
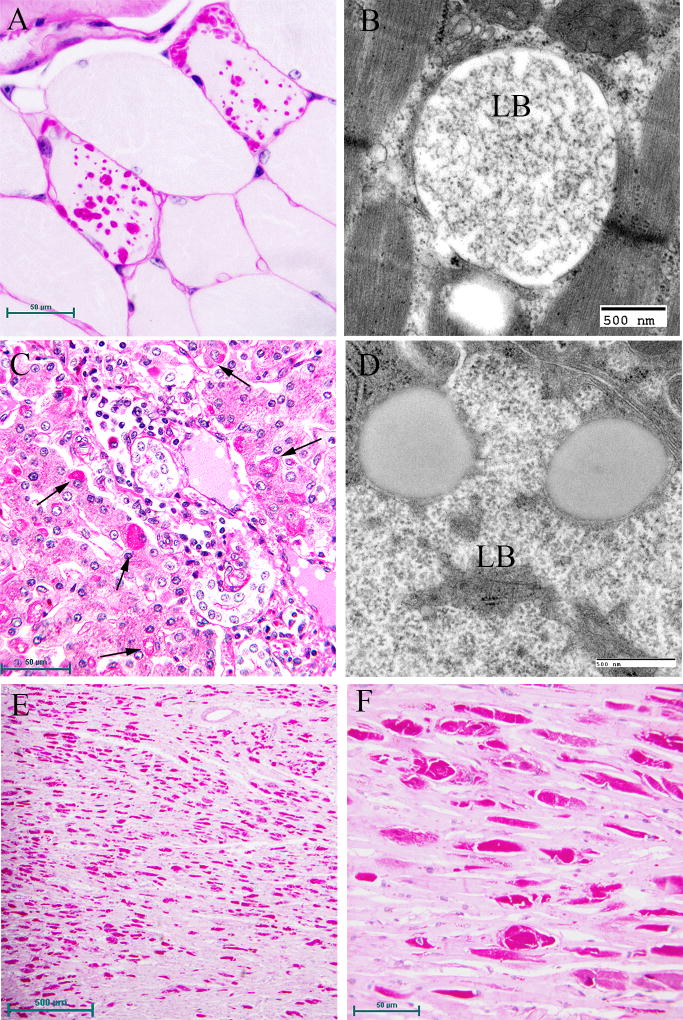
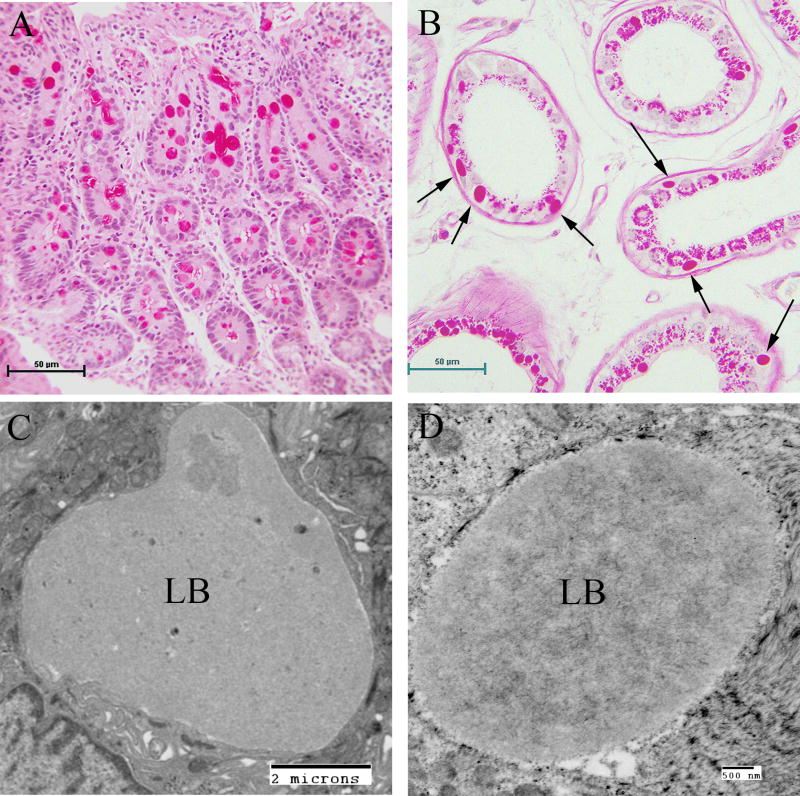
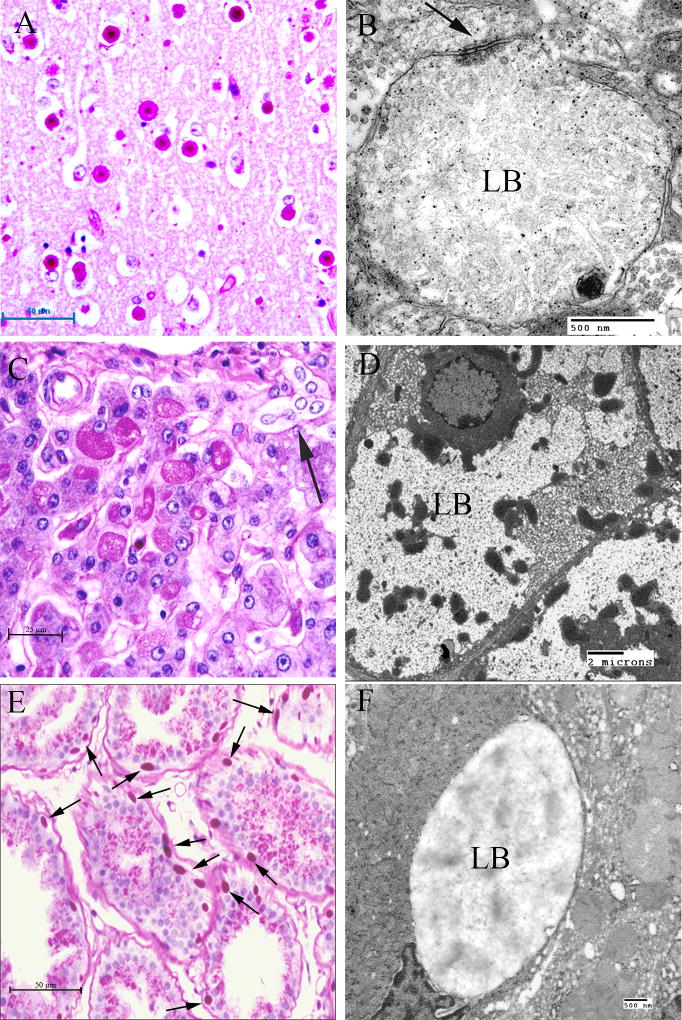
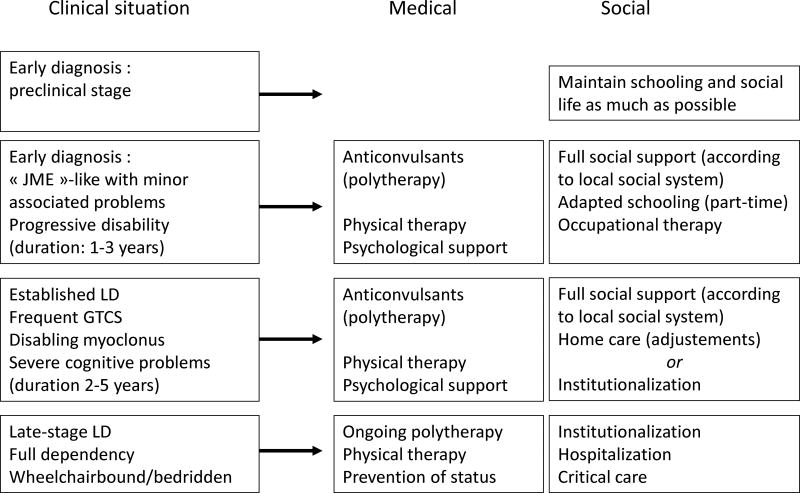
Lafora disease (LD) is an autosomal recessive progressive myoclonus epilepsy due to mutations in the EPM2A (laforin) and EPM2B (malin) genes, with no substantial genotype-phenotype differences between the two. Founder effects and recurrent mutations are common, and mostly isolated to specific ethnic groups and/or geographical locations. Pathologically, LD is characterized by distinctive polyglucosans, which are formations of abnormal glycogen. Polyglucosans, or Lafora bodies (LB) are typically found in the brain, periportal hepatocytes of the liver, skeletal and cardiac myocytes, and in the eccrine duct and apocrine myoepithelial cells of sweat glands. Mouse models of the disease and other naturally occurring animal models have similar pathology and phenotype. Hypotheses of LB formation remain controversial, with compelling evidence and caveats for each hypothesis. However, it is clear that the laforin and malin functions regulating glycogen structure are key. With the exception of a few missense mutations LD is clinically homogeneous, with onset in adolescence. Symptoms begin with seizures, and neurological decline follows soon after. The disease course is progressive and fatal, with death occurring within 10 years of onset. Antiepileptic drugs are mostly non-effective, with none having a major influence on the progression of cognitive and behavioral symptoms. Diagnosis and genetic counseling are important aspects of LD, and social support is essential in disease management. Future therapeutics for LD will revolve around the pathogenesics of the disease. Currently, efforts at identifying compounds or approaches to reduce brain glycogen synthesis appear to be highly promising.
Keywords: EPM2A; EPM2B; Lafora; glycogen phosphatase; laforin; malin; progressive myoclonus epilepsies; ubiquitin.
Figures

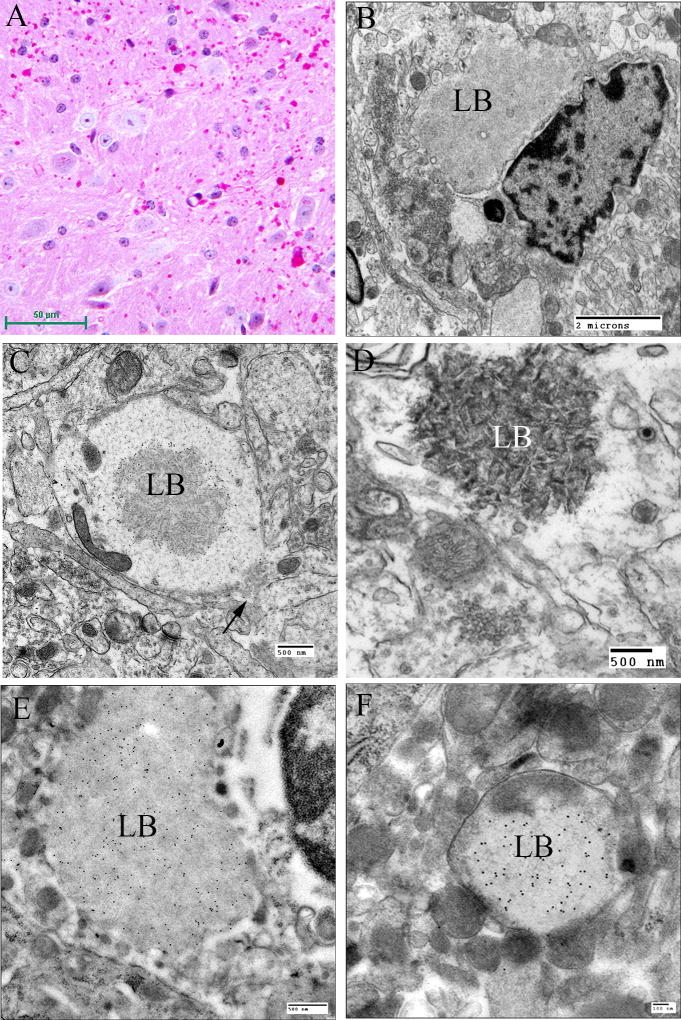
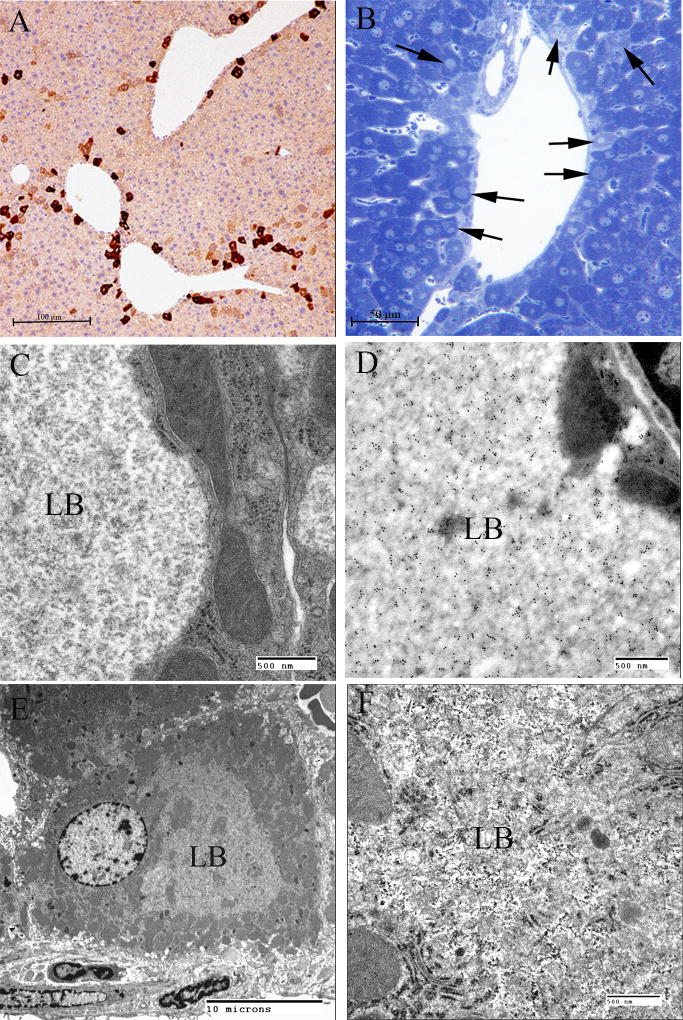
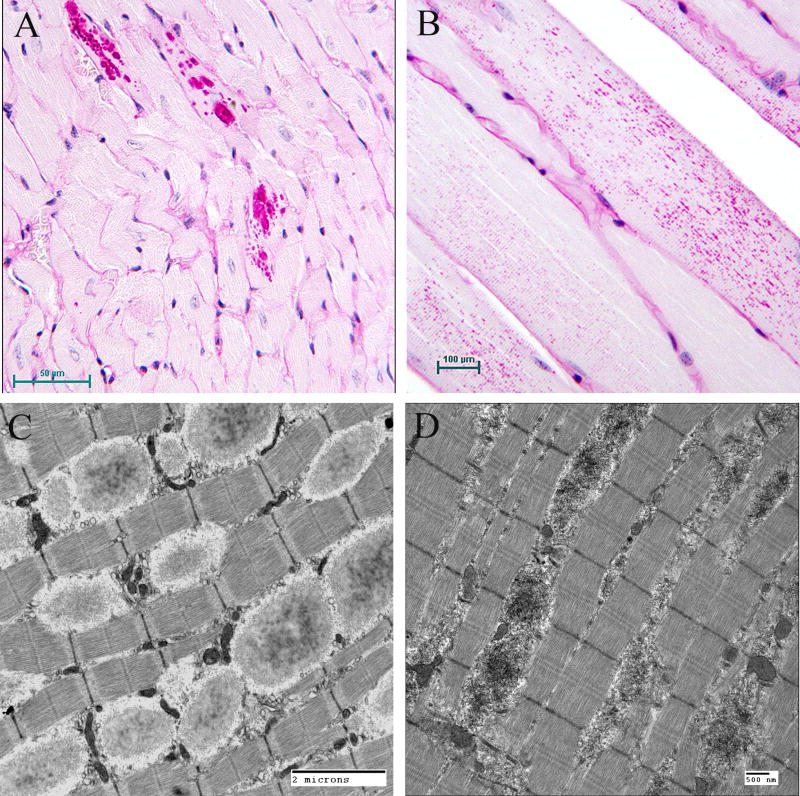
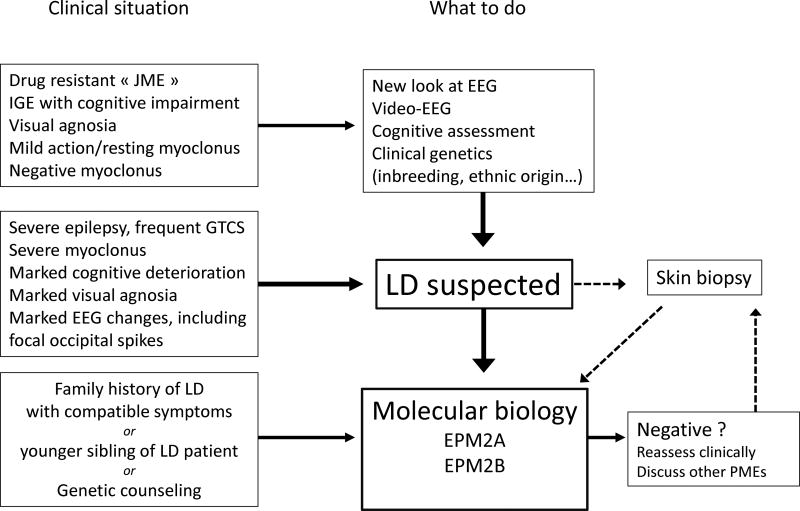
References
-
- Andrade DM, Ackerley CA, Minett TS, et al. Skin biopsy in Lafora disease: genotype-phenotype correlations and diagnostic pitfalls. Neurology. 2003;61:611–614. - PubMed
-
- Andrade DM, del Campo JM, Moro E, Minassian BA. Nonepileptic visual hallucinations in Lafora disease. Neurology. 2005;64(7):1311–1312. - PubMed
-
- Annesi G, Sofia V, Gambardella A, et al. A novel exon 1 mutation in a patient with atypical Lafora progressive myoclonus epilepsy seen as childhood-onset cognitive deficit. Epilepsia. 2004;45:294–295. - PubMed
-
- Barbieri F, Santangelo R, Gasparo-Rippa P, Santoro M. Biopsy findings (cerebral cortex, muscle, skin) in Lafora disease. Acta Neurol. 1987;9(2):81–94. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
