

Deep sequencing of 10,000 human genomes
- PMID: 27702888
- PMCID: PMC5081584
- DOI: 10.1073/pnas.1613365113
Deep sequencing of 10,000 human genomes
Abstract
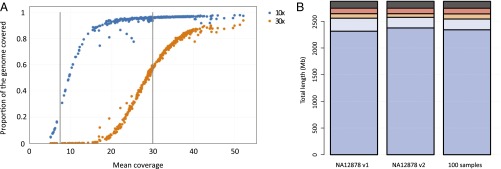
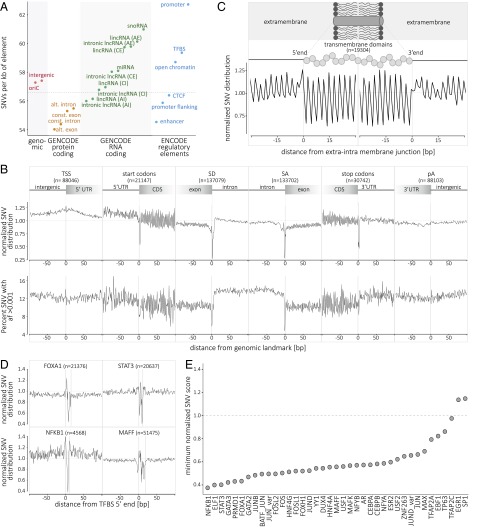
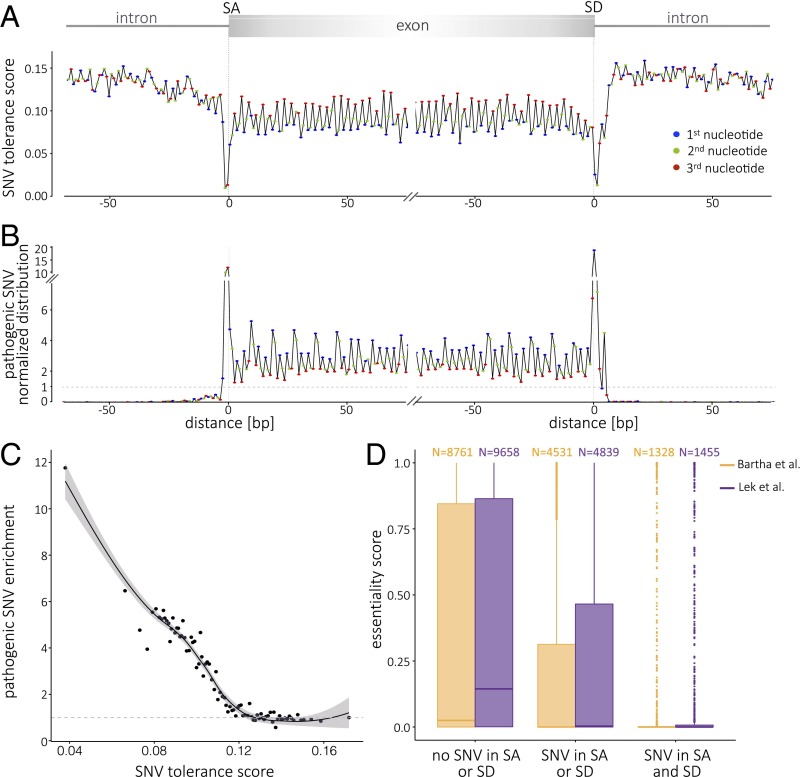
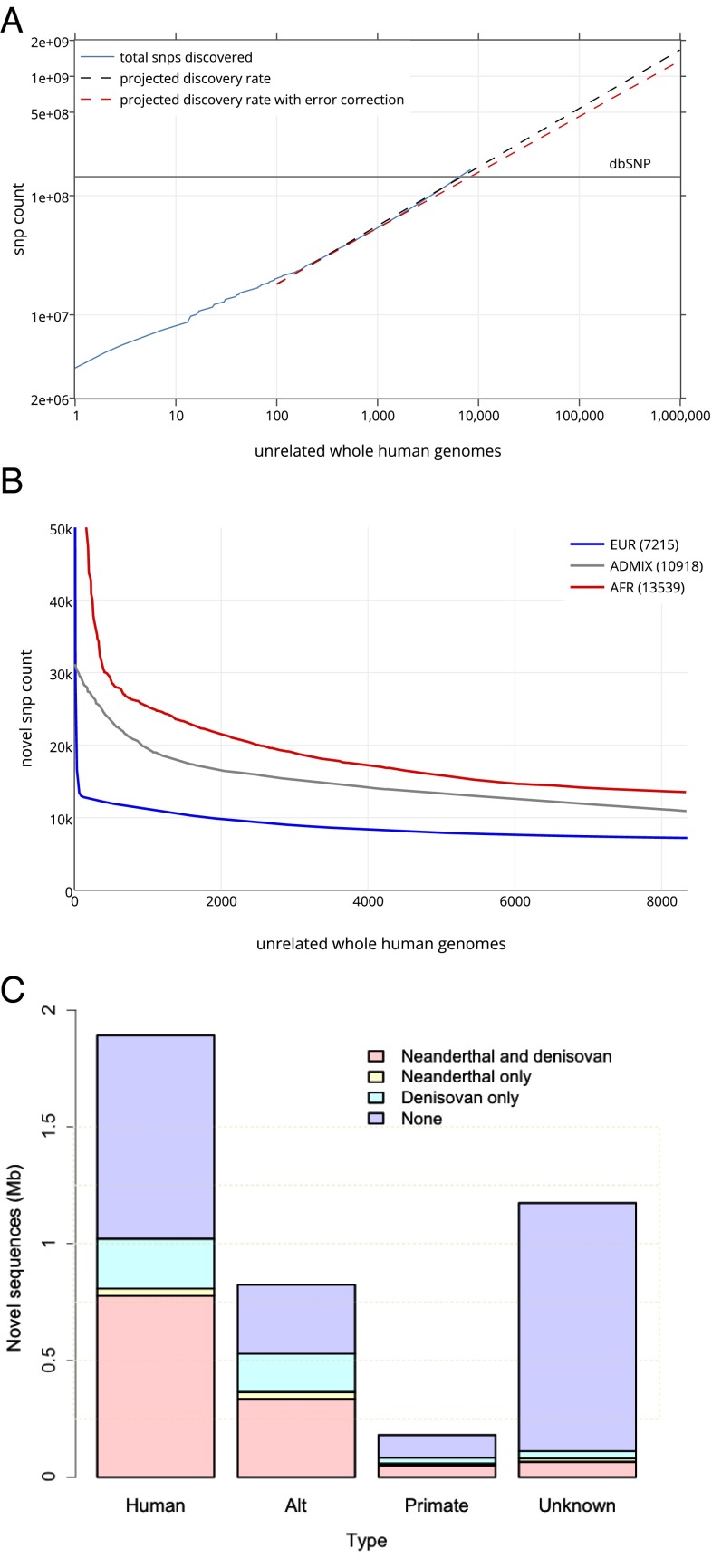
We report on the sequencing of 10,545 human genomes at 30×-40× coverage with an emphasis on quality metrics and novel variant and sequence discovery. We find that 84% of an individual human genome can be sequenced confidently. This high-confidence region includes 91.5% of exon sequence and 95.2% of known pathogenic variant positions. We present the distribution of over 150 million single-nucleotide variants in the coding and noncoding genome. Each newly sequenced genome contributes an average of 8,579 novel variants. In addition, each genome carries on average 0.7 Mb of sequence that is not found in the main build of the hg38 reference genome. The density of this catalog of variation allowed us to construct high-resolution profiles that define genomic sites that are highly intolerant of genetic variation. These results indicate that the data generated by deep genome sequencing is of the quality necessary for clinical use.
Keywords: genomics; human genetic diversity; noncoding genome.
Conflict of interest statement
The authors are employees of Human Longevity, Inc.
Figures
Comment in
-
It takes a genome to understand a village: Population scale precision medicine.Proc Natl Acad Sci U S A. 2016 Nov 1;113(44):12344-12346. doi: 10.1073/pnas.1615329113. Epub 2016 Oct 19. Proc Natl Acad Sci U S A. 2016. PMID: 27791179 Free PMC article. No abstract available.
References
-
- Genome of the Netherlands Consortium Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46(8):818–825. - PubMed
-
- Gudbjartsson DF, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet. 2015;47(5):435–444. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
