

Nat1 Deficiency Is Associated with Mitochondrial Dysfunction and Exercise Intolerance in Mice
- PMID: 27705799
- PMCID: PMC5097870
- DOI: 10.1016/j.celrep.2016.09.005
Nat1 Deficiency Is Associated with Mitochondrial Dysfunction and Exercise Intolerance in Mice
Abstract
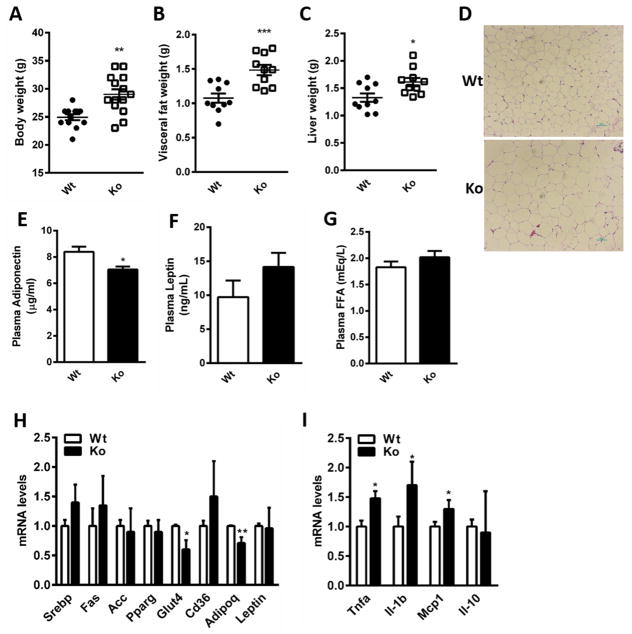
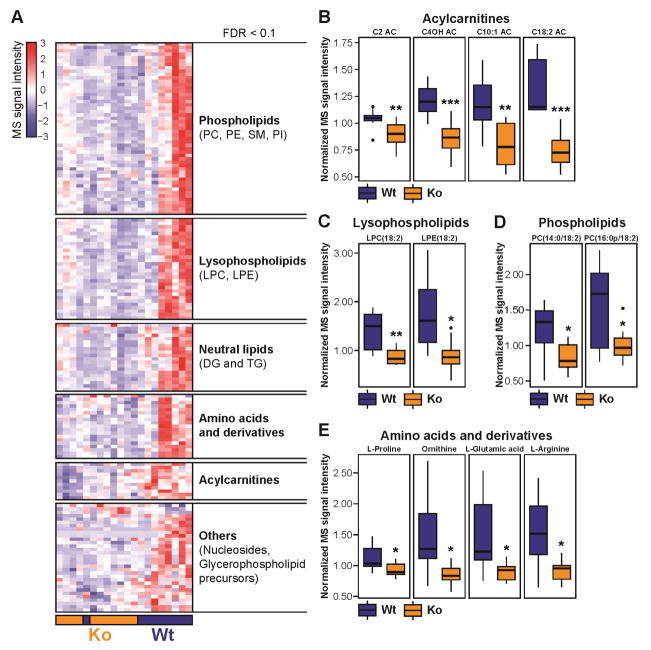
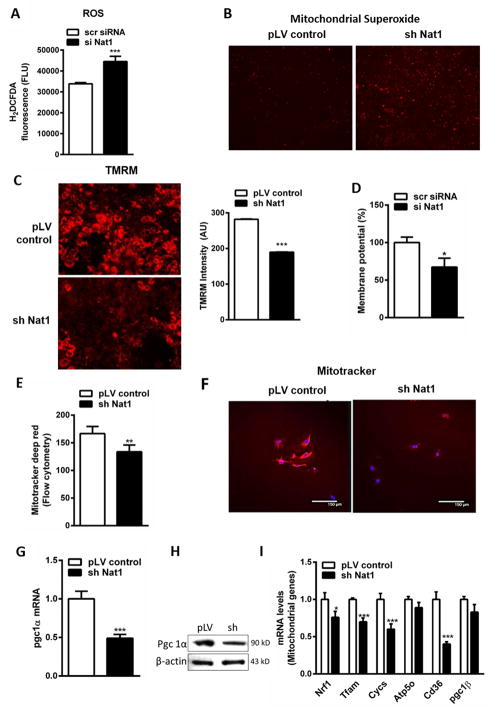
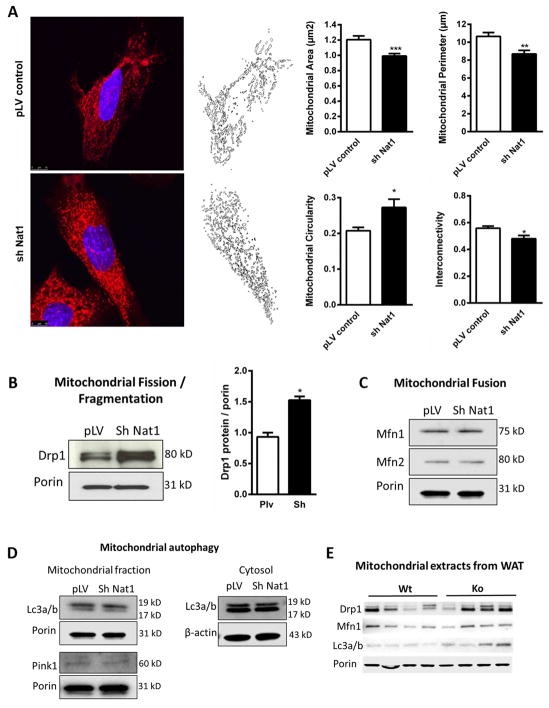
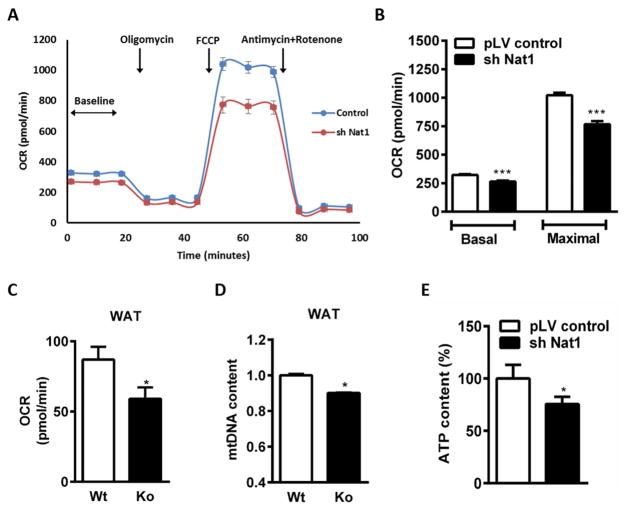
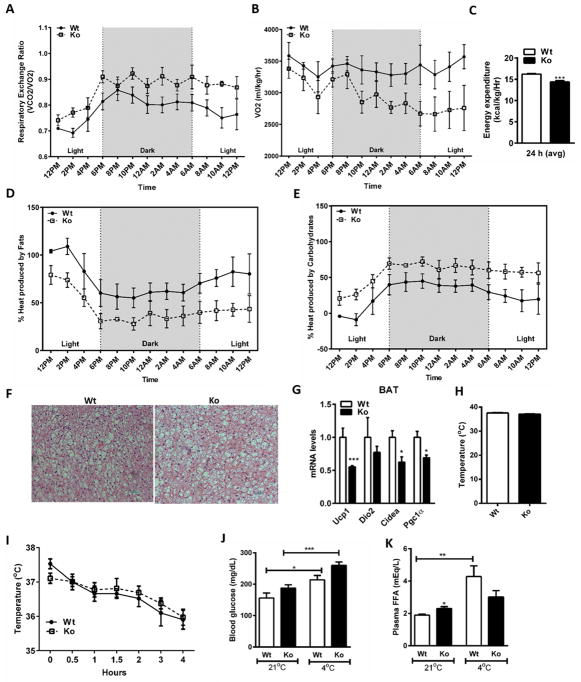
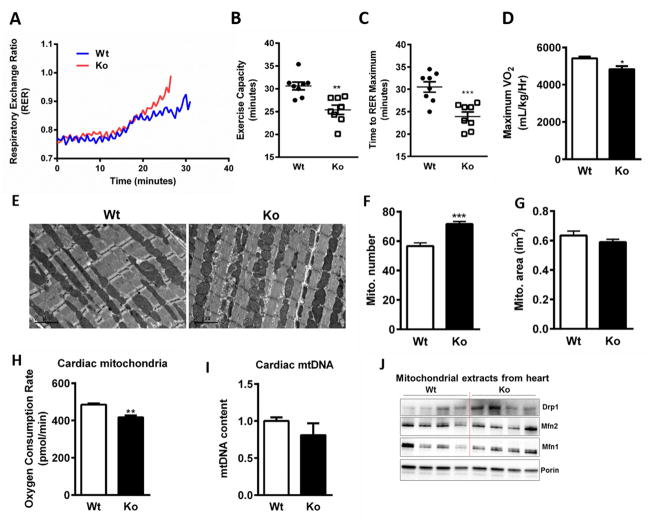
We recently identified human N-acetyltransferase 2 (NAT2) as an insulin resistance (IR) gene. Here, we examine the cellular mechanism linking NAT2 to IR and find that Nat1 (mouse ortholog of NAT2) is co-regulated with key mitochondrial genes. RNAi-mediated silencing of Nat1 led to mitochondrial dysfunction characterized by increased intracellular reactive oxygen species and mitochondrial fragmentation as well as decreased mitochondrial membrane potential, biogenesis, mass, cellular respiration, and ATP generation. These effects were consistent in 3T3-L1 adipocytes, C2C12 myoblasts, and in tissues from Nat1-deficient mice, including white adipose tissue, heart, and skeletal muscle. Nat1-deficient mice had changes in plasma metabolites and lipids consistent with a decreased ability to utilize fats for energy and a decrease in basal metabolic rate and exercise capacity without altered thermogenesis. Collectively, our results suggest that Nat1 deficiency results in mitochondrial dysfunction, which may constitute a mechanistic link between this gene and IR.
Keywords: NAT2; Nat1; adipose tissue; basal metabolic rate; fatty acids; insulin resistance; mitochondria; mitochondrial dysfunction; reactive oxygen species.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Bogacka I, Ukropcova B, McNeil M, Gimble JM, Smith SR. Structural and functional consequences of mitochondrial biogenesis in human adipocytes in vitro. J Clin Endocrinol Metab. 2005a;90:6650–6656. - PubMed
-
- Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005b;54:1392–1399. - PubMed
-
- Cannon B, Nedergaard J. Nonshivering thermogenesis and its adequate measurement in metabolic studies. Journal of Experimental Biology. 2011;214:242–253. - PubMed
-
- Choo HJ, Kim JH, Kwon OB, Lee CS, Mun JY, Han SS, Yoon YS, Yoon G, Choi KM, Ko YG. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia. 2006;49:784–791. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
