

A high-quality human reference panel reveals the complexity and distribution of genomic structural variants
- PMID: 27708267
- PMCID: PMC5059695
- DOI: 10.1038/ncomms12989
A high-quality human reference panel reveals the complexity and distribution of genomic structural variants
Abstract
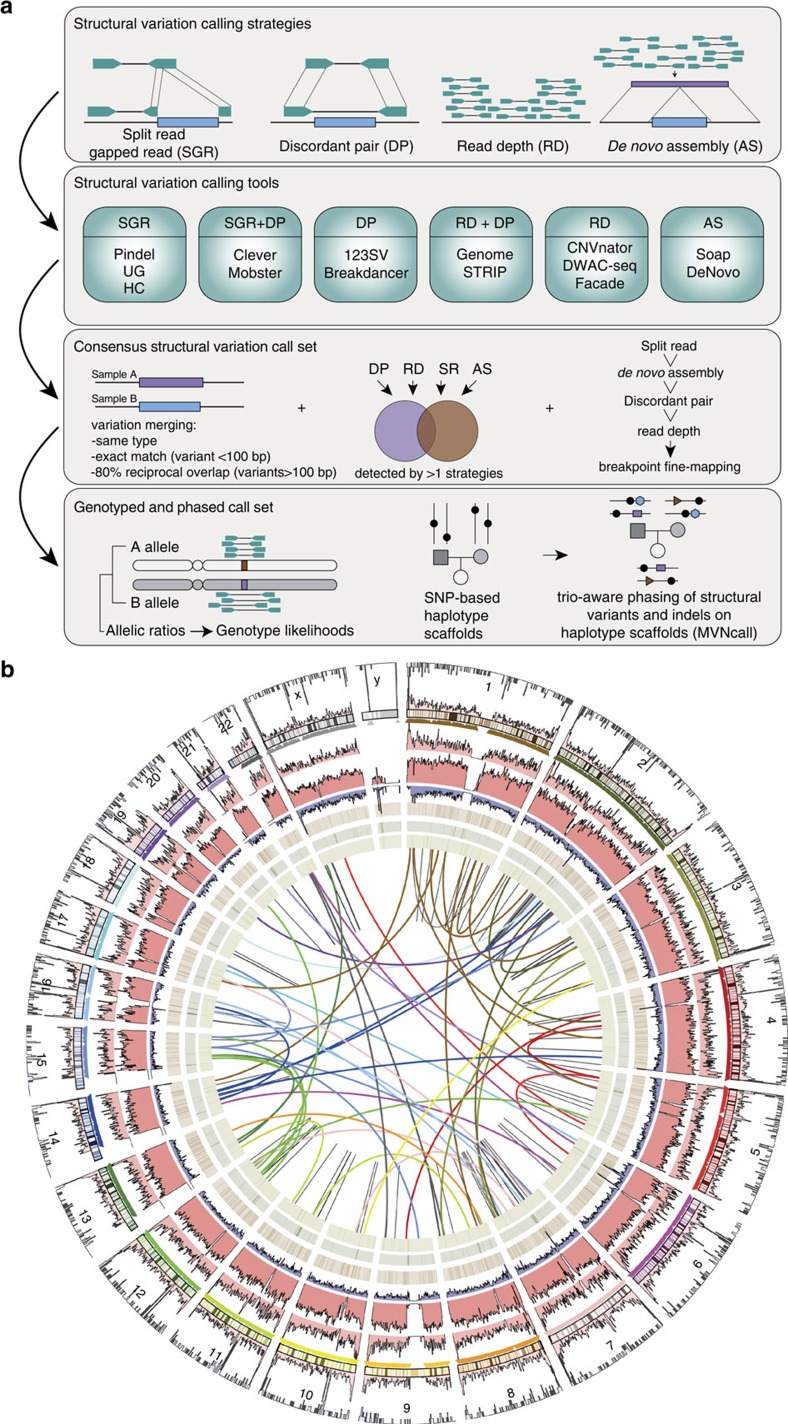
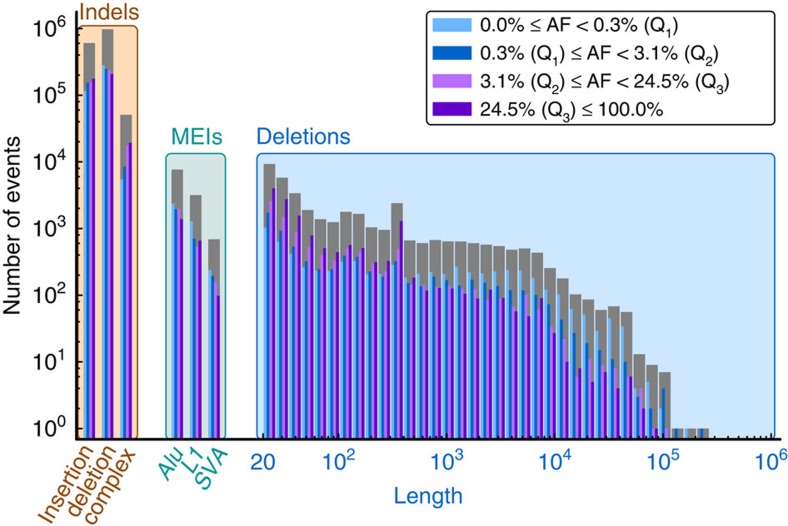
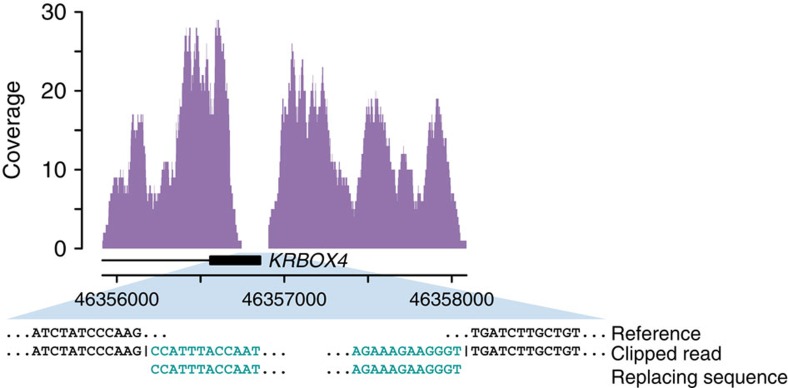
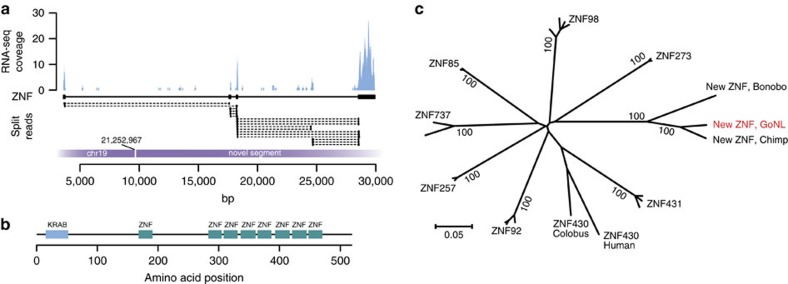
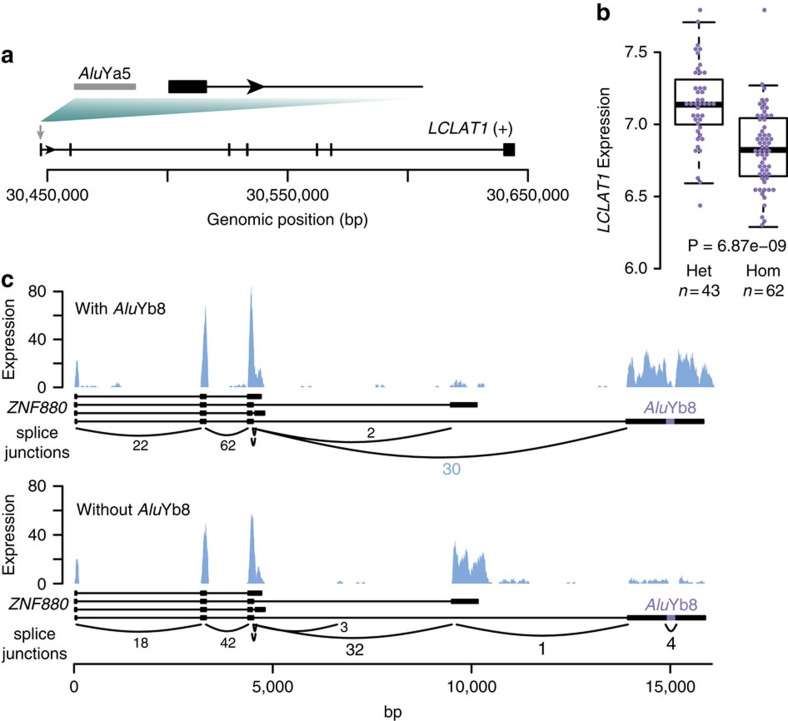
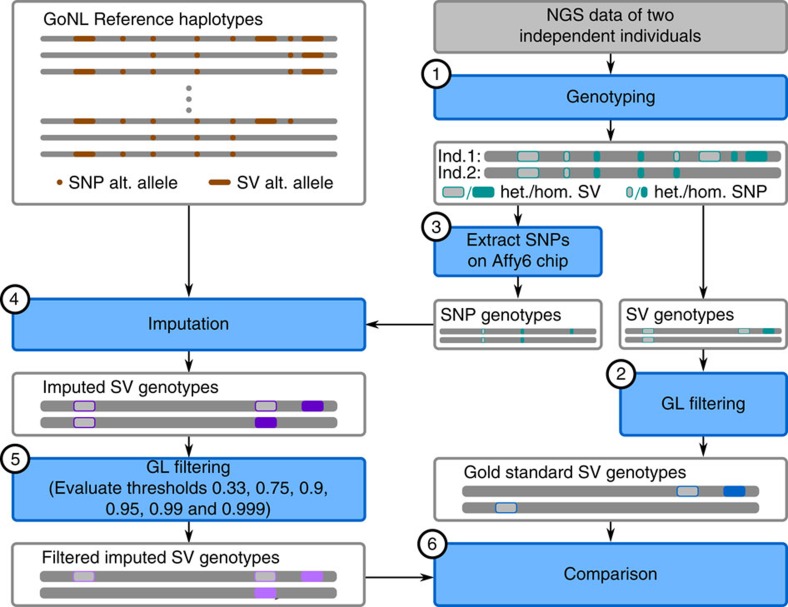
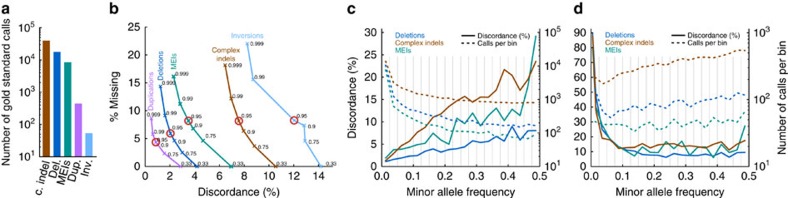
Structural variation (SV) represents a major source of differences between individual human genomes and has been linked to disease phenotypes. However, the majority of studies provide neither a global view of the full spectrum of these variants nor integrate them into reference panels of genetic variation. Here, we analyse whole genome sequencing data of 769 individuals from 250 Dutch families, and provide a haplotype-resolved map of 1.9 million genome variants across 9 different variant classes, including novel forms of complex indels, and retrotransposition-mediated insertions of mobile elements and processed RNAs. A large proportion are previously under reported variants sized between 21 and 100 bp. We detect 4 megabases of novel sequence, encoding 11 new transcripts. Finally, we show 191 known, trait-associated SNPs to be in strong linkage disequilibrium with SVs and demonstrate that our panel facilitates accurate imputation of SVs in unrelated individuals.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
