

An efficient algorithm for protein structure comparison using elastic shape analysis
- PMID: 27708689
- PMCID: PMC5041553
- DOI: 10.1186/s13015-016-0089-1
An efficient algorithm for protein structure comparison using elastic shape analysis
Abstract
Background: Protein structure comparison play important role in in silico functional prediction of a new protein. It is also used for understanding the evolutionary relationships among proteins. A variety of methods have been proposed in literature for comparing protein structures but they have their own limitations in terms of accuracy and complexity with respect to computational time and space. There is a need to improve the computational complexity in comparison/alignment of proteins through incorporation of important biological and structural properties in the existing techniques.
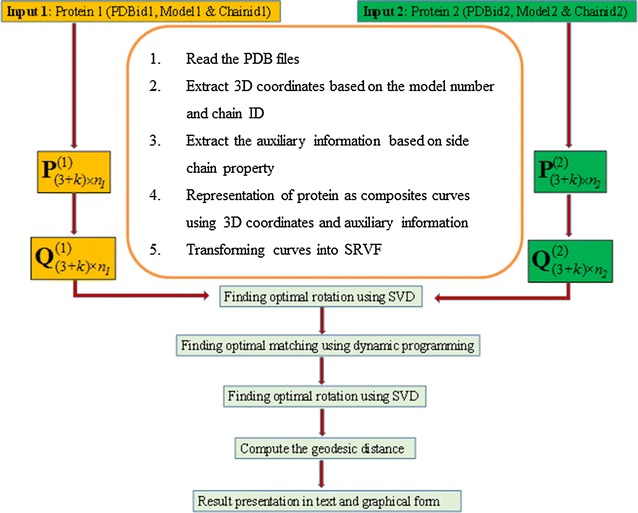
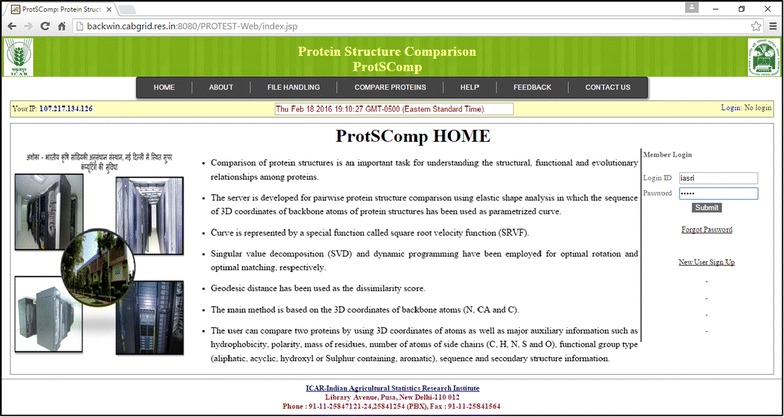
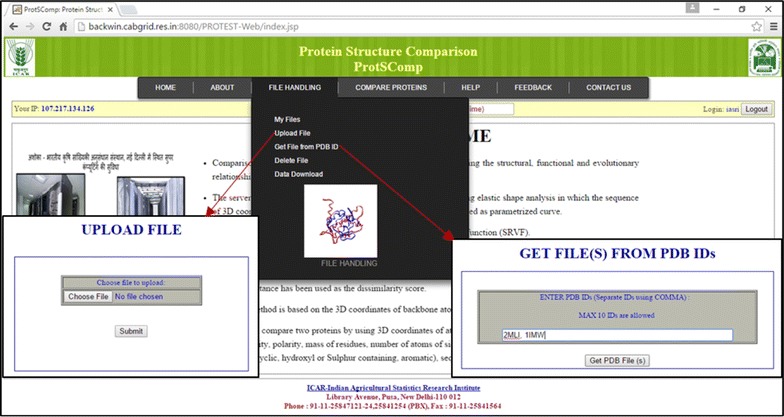
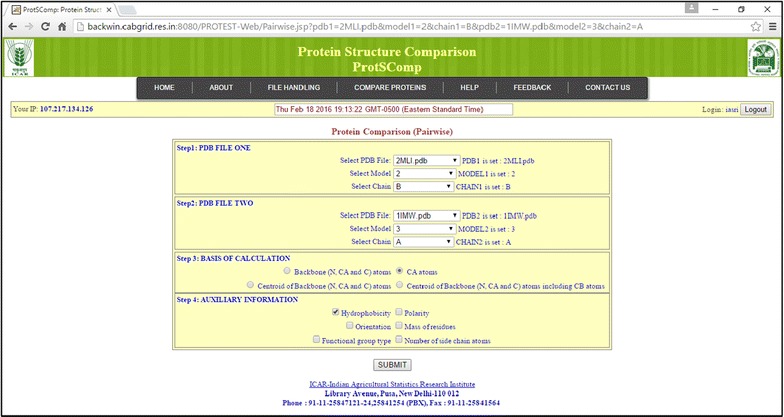
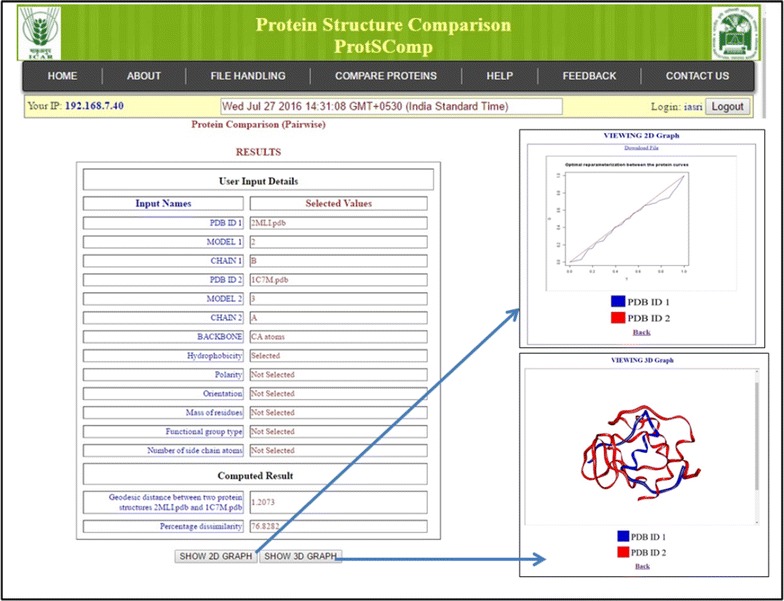
Results: An efficient algorithm has been developed for comparing protein structures using elastic shape analysis in which the sequence of 3D coordinates atoms of protein structures supplemented by additional auxiliary information from side-chain properties are incorporated. The protein structure is represented by a special function called square-root velocity function. Furthermore, singular value decomposition and dynamic programming have been employed for optimal rotation and optimal matching of the proteins, respectively. Also, geodesic distance has been calculated and used as the dissimilarity score between two protein structures. The performance of the developed algorithm is tested and found to be more efficient, i.e., running time reduced by 80-90 % without compromising accuracy of comparison when compared with the existing methods. Source codes for different functions have been developed in R. Also, user friendly web-based application called ProtSComp has been developed using above algorithm for comparing protein 3D structures and is accessible free.
Conclusions: The methodology and algorithm developed in this study is taking considerably less computational time without loss of accuracy (Table 2). The proposed algorithm is considering different criteria of representing protein structures using 3D coordinates of atoms and inclusion of residue wise molecular properties as auxiliary information.
Keywords: Backbone atoms; Geodesic distance; Protein structure comparison; Side chain properties.
Figures
Similar articles
-
Efficient dynamic programming algorithm with prior knowledge for protein β-strand alignment.J Theor Biol. 2017 Mar 21;417:43-50. doi: 10.1016/j.jtbi.2017.01.018. Epub 2017 Jan 18. J Theor Biol. 2017. PMID: 28108305
-
Structural alignment of protein descriptors - a combinatorial model.BMC Bioinformatics. 2016 Sep 17;17:383. doi: 10.1186/s12859-016-1237-9. BMC Bioinformatics. 2016. PMID: 27639380 Free PMC article.
-
Revealing divergent evolution, identifying circular permutations and detecting active-sites by protein structure comparison.BMC Struct Biol. 2006 Sep 2;6:18. doi: 10.1186/1472-6807-6-18. BMC Struct Biol. 2006. PMID: 16948858 Free PMC article.
-
Comparing protein structures and inferring functions with a novel three-dimensional Yau-Hausdorff method.J Biomol Struct Dyn. 2019 Oct;37(16):4151-4160. doi: 10.1080/07391102.2018.1540359. Epub 2018 Dec 5. J Biomol Struct Dyn. 2019. PMID: 30518311
-
Combining evolutionary and structural information for local protein structure prediction.Proteins. 2004 Sep 1;56(4):782-94. doi: 10.1002/prot.20158. Proteins. 2004. PMID: 15281130
Cited by
-
Automated shape-based clustering of 3D immunoglobulin protein structures in chronic lymphocytic leukemia.BMC Bioinformatics. 2018 Nov 20;19(Suppl 14):414. doi: 10.1186/s12859-018-2381-1. BMC Bioinformatics. 2018. PMID: 30453883 Free PMC article.
-
A review of visualisations of protein fold networks and their relationship with sequence and function.Biol Rev Camb Philos Soc. 2023 Feb;98(1):243-262. doi: 10.1111/brv.12905. Epub 2022 Oct 9. Biol Rev Camb Philos Soc. 2023. PMID: 36210328 Free PMC article. Review.
-
Image-based effective feature generation for protein structural class and ligand binding prediction.PeerJ Comput Sci. 2020 Feb 3;6:e253. doi: 10.7717/peerj-cs.253. eCollection 2020. PeerJ Comput Sci. 2020. PMID: 33816905 Free PMC article.
-
Simplifying Transforms for General Elastic Metrics on the Space of Plane Curves.SIAM J Imaging Sci. 2020;13(1):445-473. doi: 10.1137/19m1265132. Epub 2020 Mar 12. SIAM J Imaging Sci. 2020. PMID: 34386150 Free PMC article.
-
Genetic Code Optimization for Cotranslational Protein Folding: Codon Directional Asymmetry Correlates with Antiparallel Betasheets, tRNA Synthetase Classes.Comput Struct Biotechnol J. 2017 Aug 12;15:412-424. doi: 10.1016/j.csbj.2017.08.001. eCollection 2017. Comput Struct Biotechnol J. 2017. PMID: 28924459 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
