

Sphingosine 1-Phosphate Activation of EGFR As a Novel Target for Meningitic Escherichia coli Penetration of the Blood-Brain Barrier
- PMID: 27711202
- PMCID: PMC5053521
- DOI: 10.1371/journal.ppat.1005926
Sphingosine 1-Phosphate Activation of EGFR As a Novel Target for Meningitic Escherichia coli Penetration of the Blood-Brain Barrier
Abstract
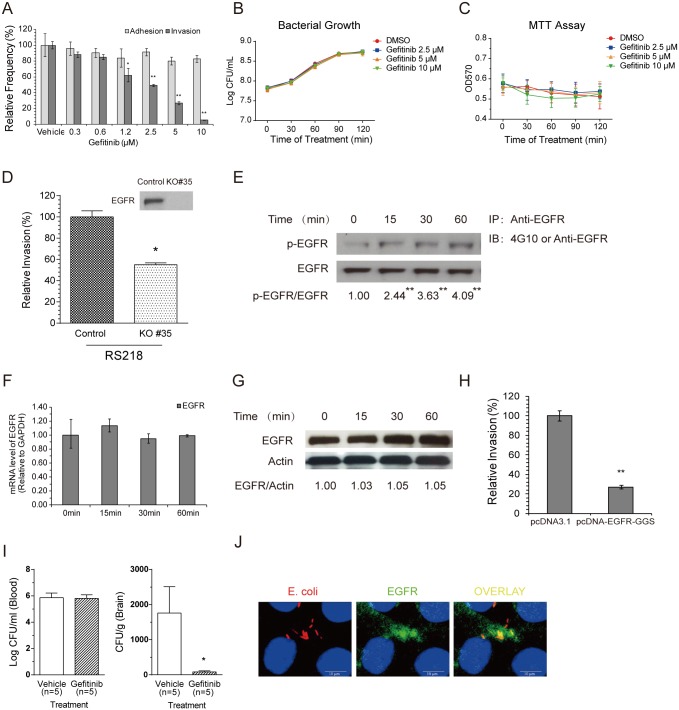
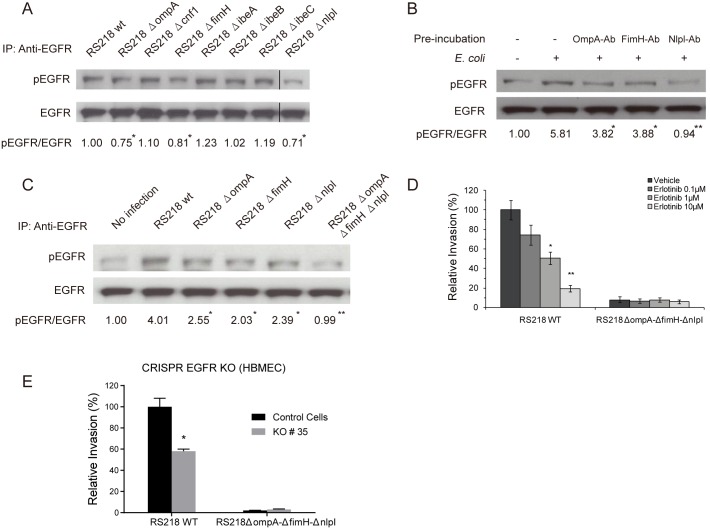
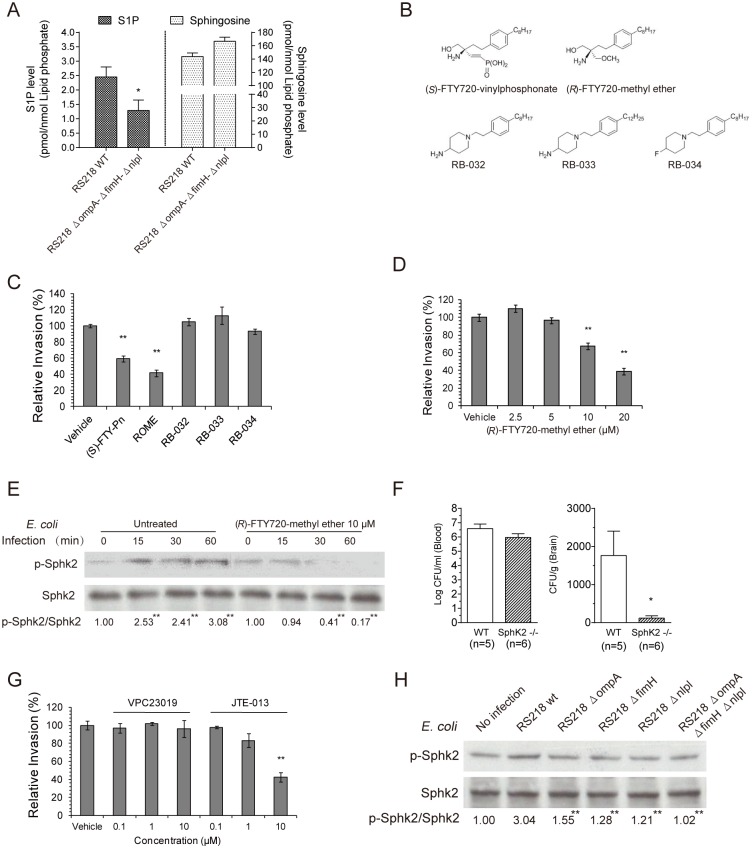
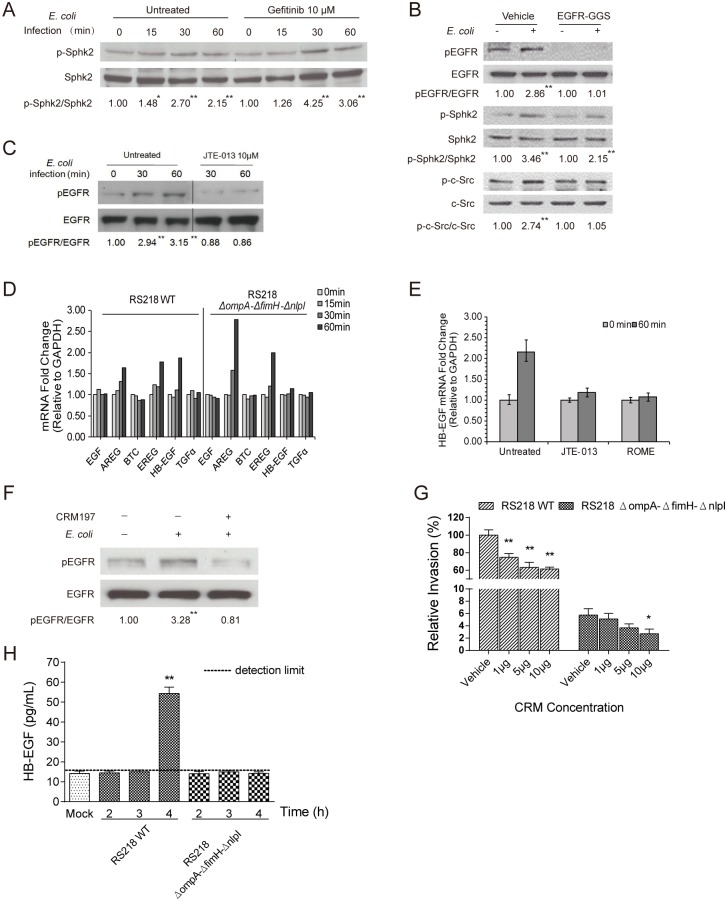
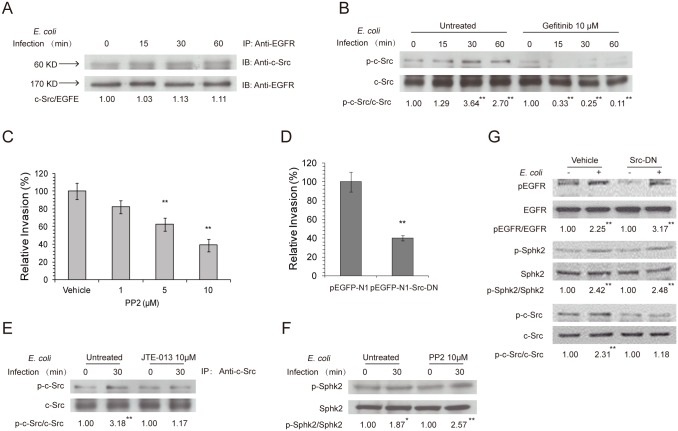
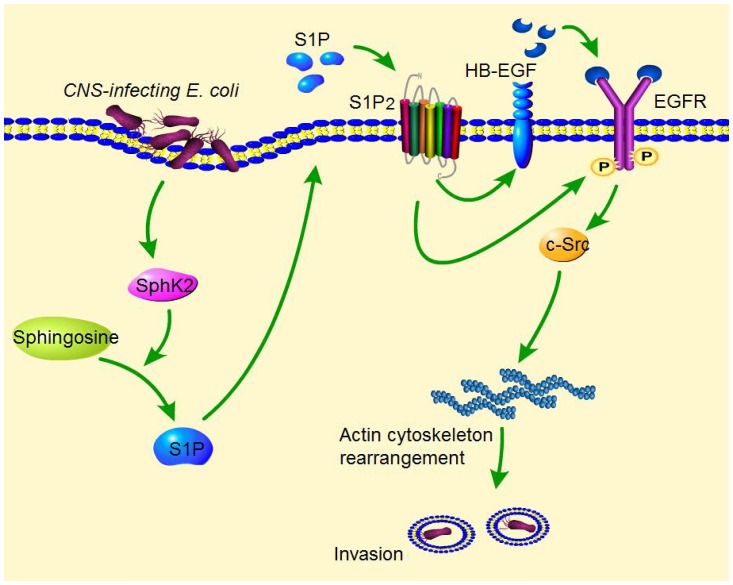
Central nervous system (CNS) infection continues to be an important cause of mortality and morbidity, necessitating new approaches for investigating its pathogenesis, prevention and therapy. Escherichia coli is the most common Gram-negative bacillary organism causing meningitis, which develops following penetration of the blood-brain barrier (BBB). By chemical library screening, we identified epidermal growth factor receptor (EGFR) as a contributor to E. coli invasion of the BBB in vitro. Here, we obtained the direct evidence that CNS-infecting E. coli exploited sphingosine 1-phosphate (S1P) for EGFR activation in penetration of the BBB in vitro and in vivo. We found that S1P was upstream of EGFR and participated in EGFR activation through S1P receptor as well as through S1P-mediated up-regulation of EGFR-related ligand HB-EGF, and blockade of S1P function through targeting sphingosine kinase and S1P receptor inhibited EGFR activation, and also E. coli invasion of the BBB. We further found that both S1P and EGFR activations occurred in response to the same E. coli proteins (OmpA, FimH, NlpI), and that S1P and EGFR promoted E. coli invasion of the BBB by activating the downstream c-Src. These findings indicate that S1P and EGFR represent the novel host targets for meningitic E. coli penetration of the BBB, and counteracting such targets provide a novel approach for controlling E. coli meningitis in the era of increasing resistance to conventional antibiotics.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
