

Selinexor, a Selective Inhibitor of Nuclear Export (SINE) compound, acts through NF-κB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death
- PMID: 27713151
- PMCID: PMC5346685
- DOI: 10.18632/oncotarget.12428
Selinexor, a Selective Inhibitor of Nuclear Export (SINE) compound, acts through NF-κB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death
Abstract
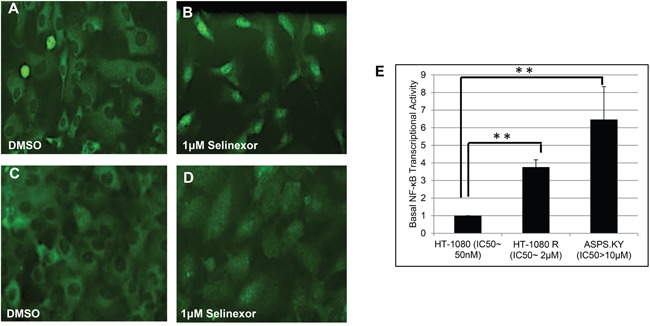
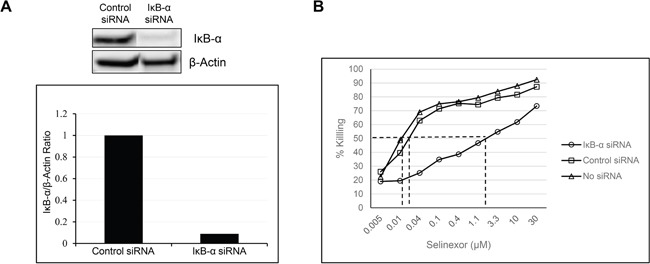
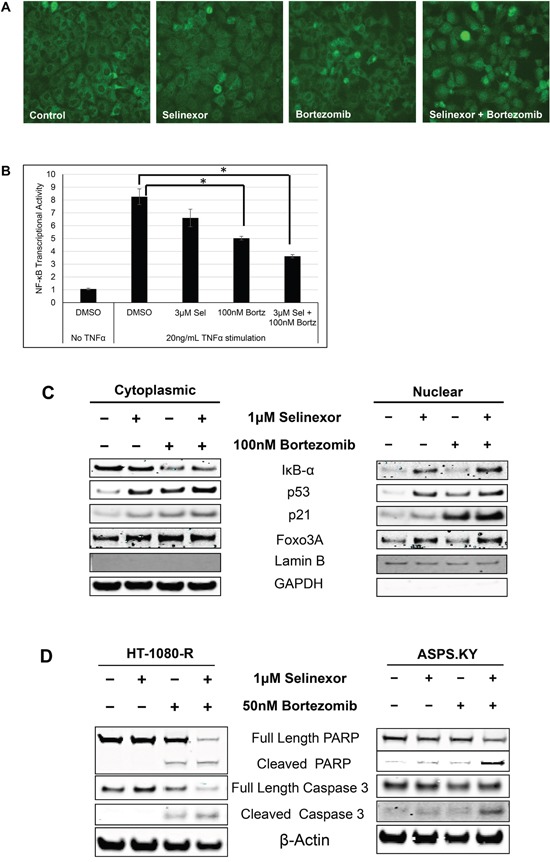
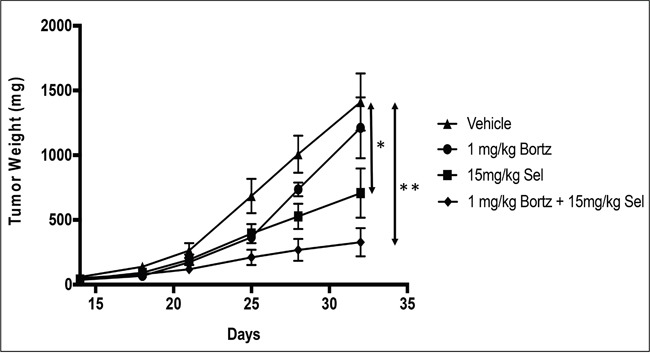
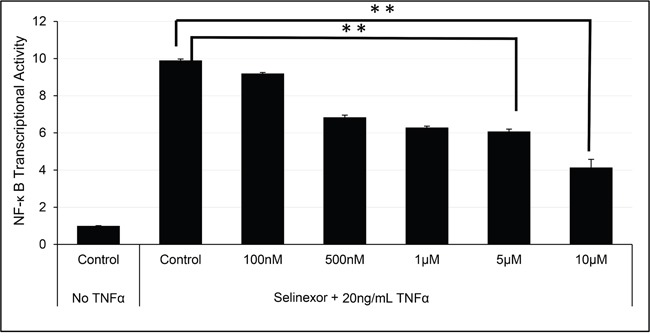
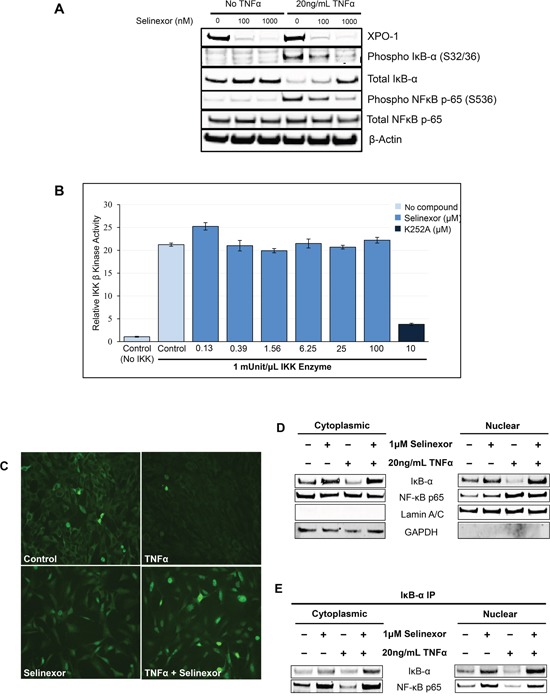
The nuclear export protein, exportin-1 (XPO1/CRM1), is overexpressed in many cancers and correlates with poor prognosis. Selinexor, a first-in-class Selective Inhibitor of Nuclear Export (SINE) compound, binds covalently to XPO1 and blocks its function. Treatment of cancer cells with selinexor results in nuclear retention of major tumor suppressor proteins and cell cycle regulators, leading to growth arrest and apoptosis. Recently, we described the selection of SINE compound resistant cells and reported elevated expression of inflammation-related genes in these cells. Here, we demonstrated that NF-κB transcriptional activity is up-regulated in cells that are naturally resistant or have acquired resistance to SINE compounds. Resistance to SINE compounds was created by knockdown of the cellular NF-κB inhibitor, IκB-α. Combination treatment of selinexor with proteasome inhibitors decreased NF-κB activity, sensitized SINE compound resistant cells and showed synergistic cytotoxicity in vitro and in vivo. Furthermore, we showed that selinexor inhibited NF-κB activity by blocking phosphorylation of the IκB-α and the NF-κB p65 subunits, protecting IκB-α from proteasome degradation and trapping IκB-α in the nucleus to suppress NF-κB activity. Therefore, combination treatment of selinexor with a proteasome inhibitor may be beneficial to patients with resistance to either single-agent.
Keywords: NF-κB; SINE; XPO1; proteasome inhibitors; selinexor.
Conflict of interest statement
TK, CA, TJU, BK, CD, WS, ML, MK, SS and YL are full time Karyopharm, Inc. employees. AA, RMM, IM, ASA are from Wayne State University School of Medicine and they have no conflicts of interest to disclose.
Figures
References
-
- Senapedis WT, Baloglu E, Landesman Y. Clinical translation of nuclear export inhibitors in cancer. Semin Cancer Biol. 2014;27:74–86. - PubMed
-
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. - PubMed
-
- Turner JG, Sullivan DM. CRM1-mediated nuclear export of proteins and drug resistance in cancer. Curr Med Chem. 2008;15:2648–2655. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
