

A method for predicting individual residue contributions to enzyme specificity and binding-site energies, and its application to MTH1
- PMID: 27714533
- PMCID: PMC5054044
- DOI: 10.1007/s00894-016-3119-5
A method for predicting individual residue contributions to enzyme specificity and binding-site energies, and its application to MTH1
Abstract
A new method for predicting the energy contributions to substrate binding and to specificity has been developed. Conventional global optimization methods do not permit the subtle effects responsible for these properties to be modeled with sufficient precision to allow confidence to be placed in the results, but by making simple alterations to the model, the precisions of the various energies involved can be improved from about ±2 kcal mol-1 to ±0.1 kcal mol-1. This technique was applied to the oxidized nucleotide pyrophosphohydrolase enzyme MTH1. MTH1 is unusual in that the binding and reaction sites are well separated-an advantage from a computational chemistry perspective, as it allows the energetics involved in docking to be modeled without the need to consider any issues relating to reaction mechanisms. In this study, two types of energy terms were investigated: the noncovalent interactions between the binding site and the substrate, and those responsible for discriminating between the oxidized nucleotide 8-oxo-dGTP and the normal dGTP. Both of these were investigated using the semiempirical method PM7 in the program MOPAC. The contributions of the individual residues to both the binding energy and the specificity of MTH1 were calculated by simulating the effect of mutations. Where comparisons were possible, all calculated results were in agreement with experimental observations. This technique provides fresh insight into the binding mechanism that enzymes use for discriminating between possible substrates.
Keywords: Binding; Docking; Enzyme specificity; MTH1; Noncovalent interactions; Nucleotide hydrolysis; PM7.
Conflict of interest statement
Compliance with ethical standards Disclaimer This work is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
Figures
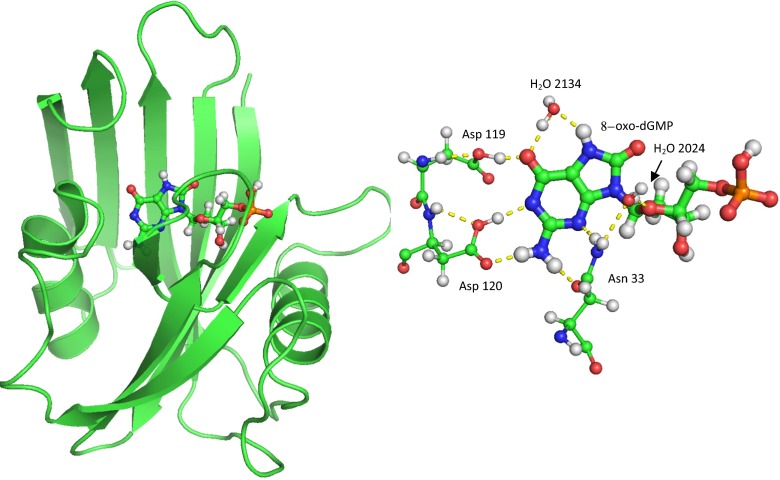
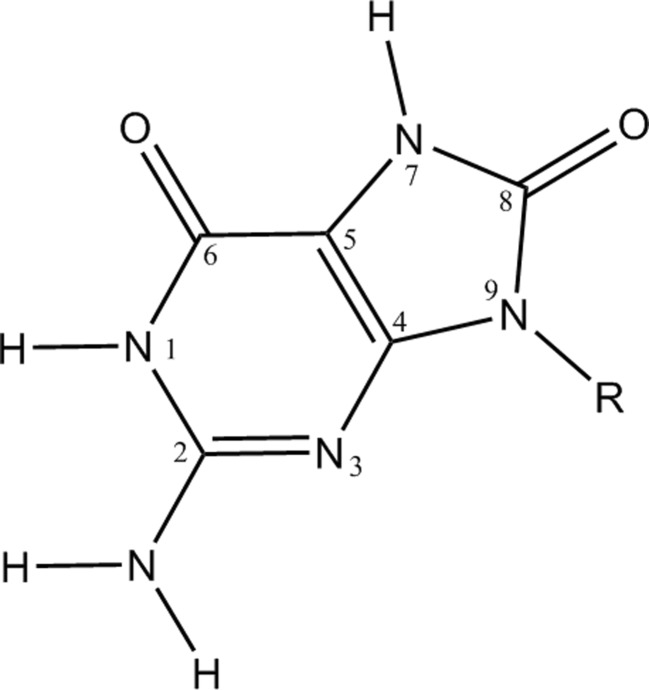
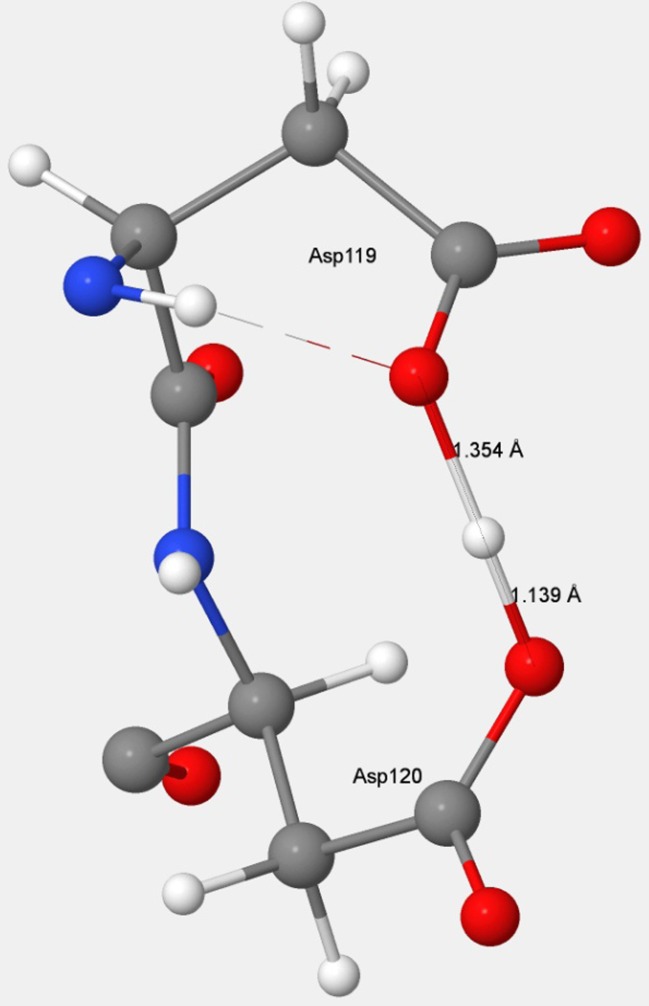
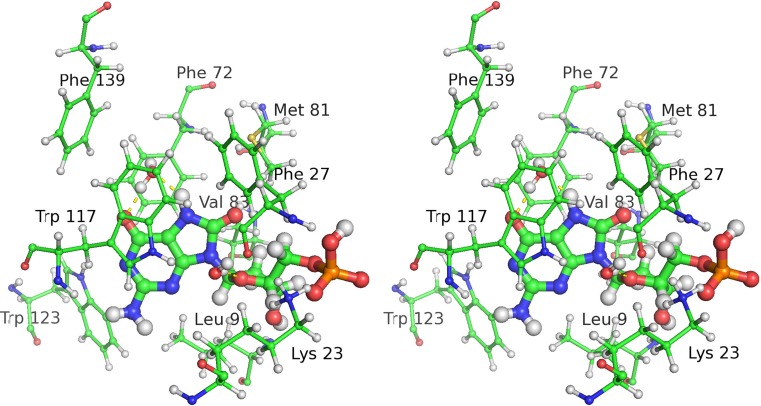
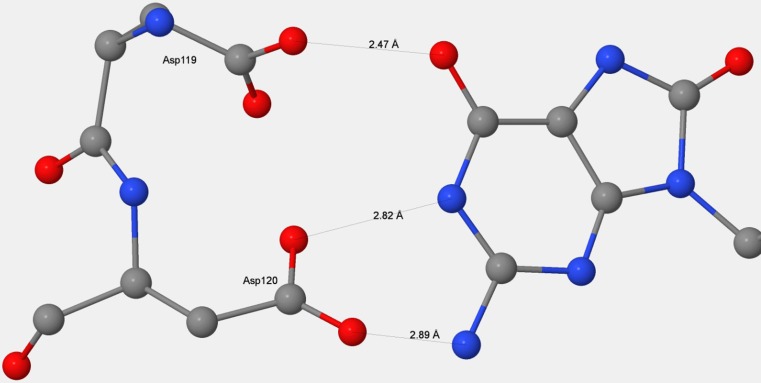
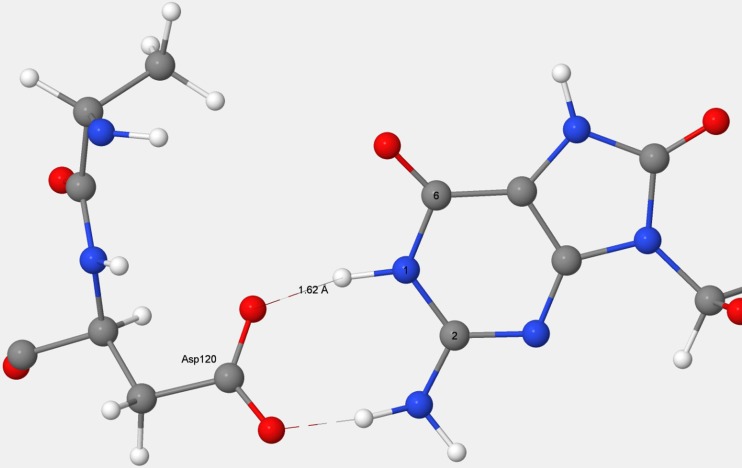
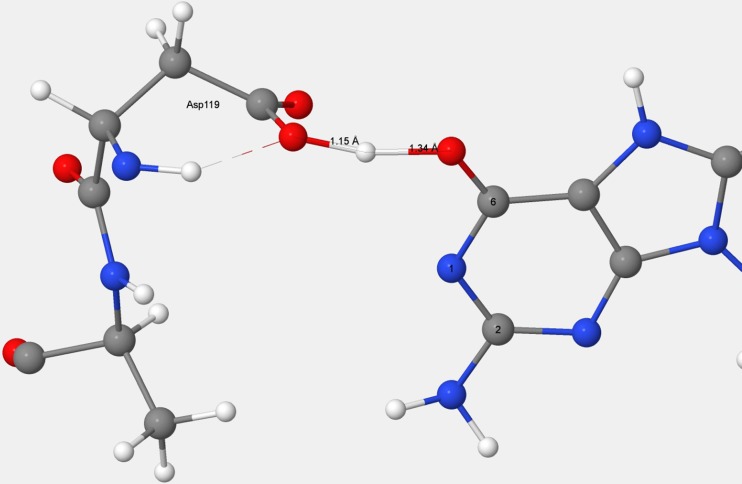
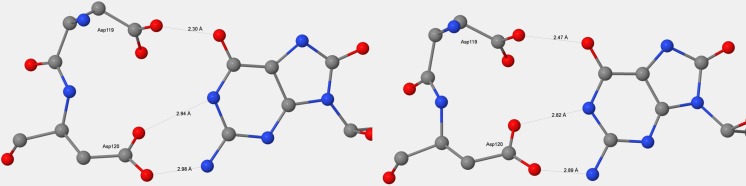
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
