

Exome and genome sequencing for inborn errors of immunity
- PMID: 27720020
- PMCID: PMC5074686
- DOI: 10.1016/j.jaci.2016.08.003
Exome and genome sequencing for inborn errors of immunity
Abstract
The advent of next-generation sequencing (NGS) in 2010 has transformed medicine, particularly the growing field of inborn errors of immunity. NGS has facilitated the discovery of novel disease-causing genes and the genetic diagnosis of patients with monogenic inborn errors of immunity. Whole-exome sequencing (WES) is presently the most cost-effective approach for research and diagnostics, although whole-genome sequencing offers several advantages. The scientific or diagnostic challenge consists in selecting 1 or 2 candidate variants among thousands of NGS calls. Variant- and gene-level computational methods, as well as immunologic hypotheses, can help narrow down this genome-wide search. The key to success is a well-informed genetic hypothesis on 3 key aspects: mode of inheritance, clinical penetrance, and genetic heterogeneity of the condition. This determines the search strategy and selection criteria for candidate alleles. Subsequent functional validation of the disease-causing effect of the candidate variant is critical. Even the most up-to-date dry lab cannot clinch this validation without a seasoned wet lab. The multifariousness of variations entails an experimental rigor even greater than traditional Sanger sequencing-based approaches in order not to assign a condition to an irrelevant variant. Finding the needle in the haystack takes patience, prudence, and discernment.
Keywords: Next-generation sequencing; primary immunodeficiency; targeted sequencing; whole-exome sequencing; whole-genome sequencing.
Copyright © 2016 American Academy of Allergy, Asthma & Immunology. Published by Elsevier Inc. All rights reserved.
Figures
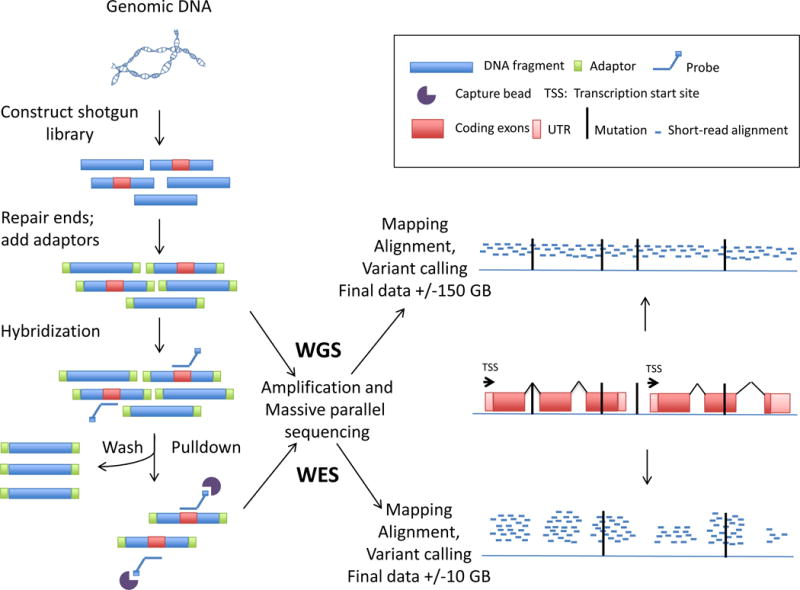
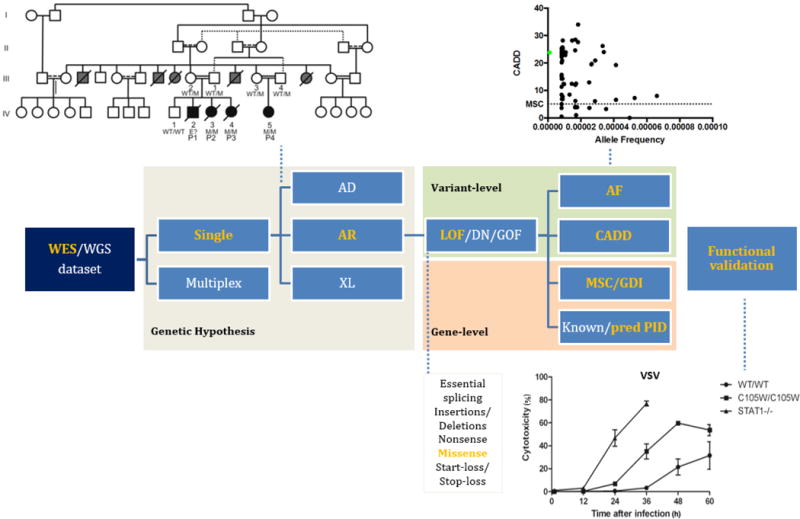
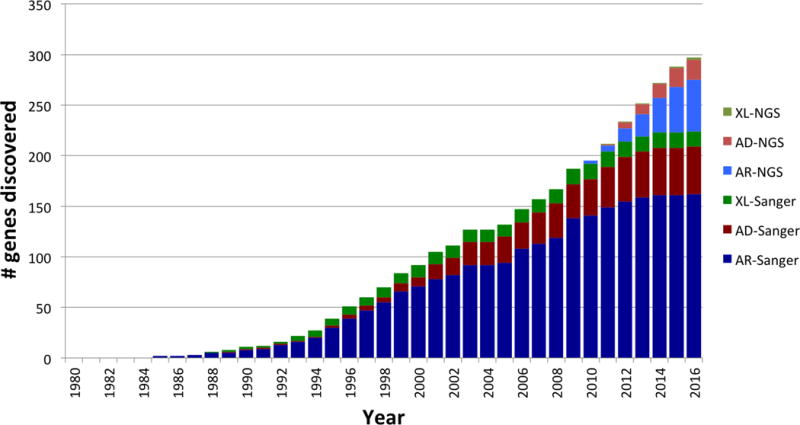
References
-
- Fang M, Abolhassani H, Lim CK, Zhang J, Hammarstrom L. Next Generation Sequencing Data Analysis in Primary Immunodeficiency Disorders – Future Directions. J Clin Immunol. 2016;36(Suppl 1):68–75. - PubMed
-
- Picard C, Fischer A. Contribution of high-throughput DNA sequencing to the study of primary immunodeficiencies. Eur J Immunol. 2014;44(10):2854–61. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
