

ETAA1 acts at stalled replication forks to maintain genome integrity
- PMID: 27723720
- PMCID: PMC5245861
- DOI: 10.1038/ncb3415
ETAA1 acts at stalled replication forks to maintain genome integrity
Abstract
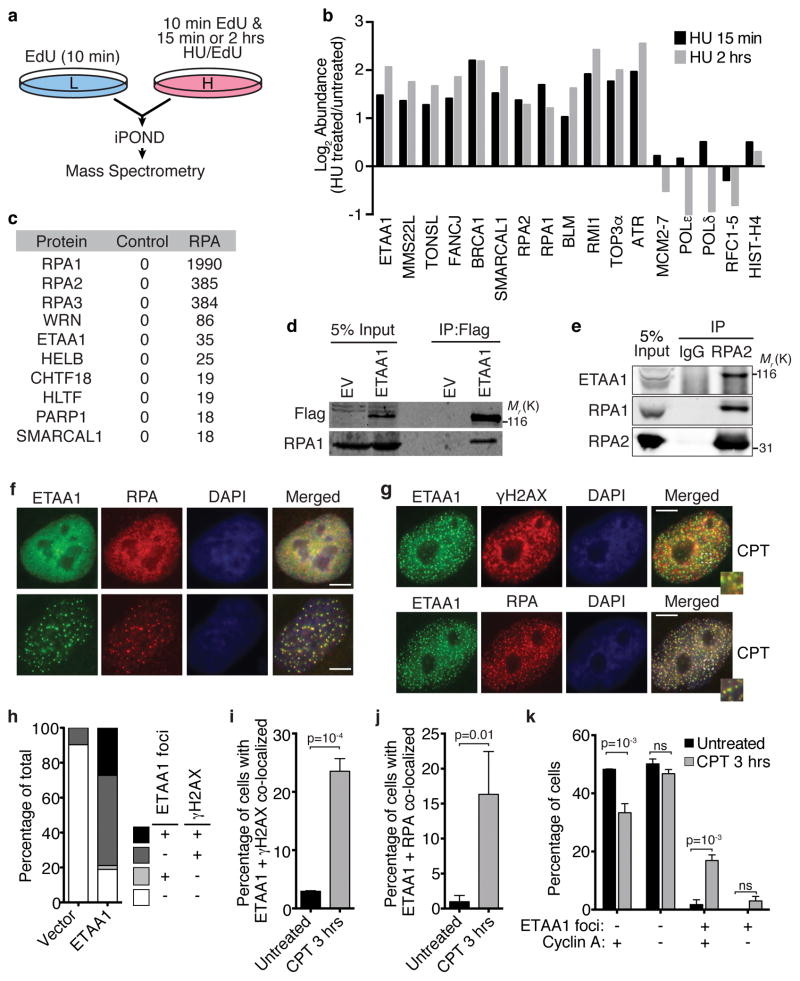
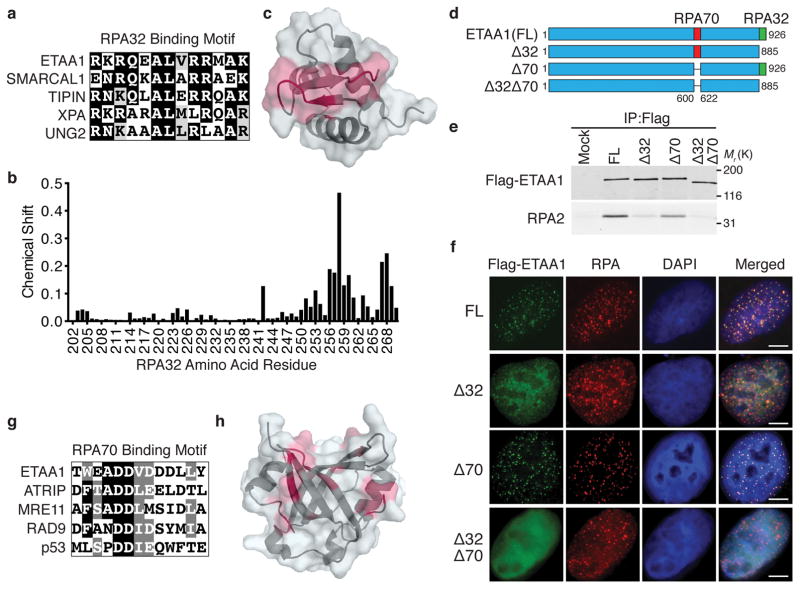
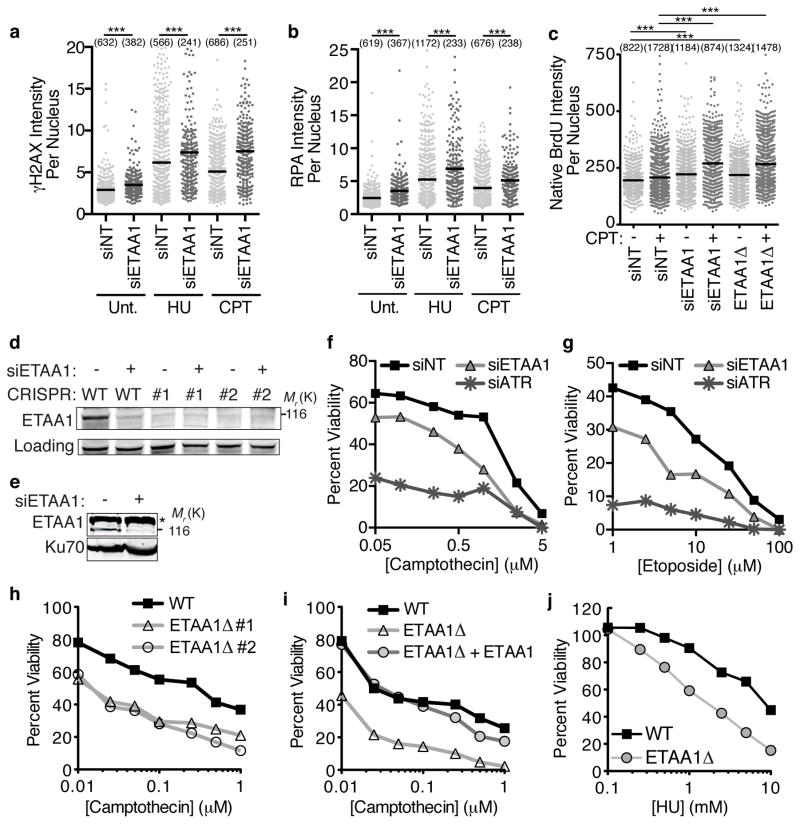
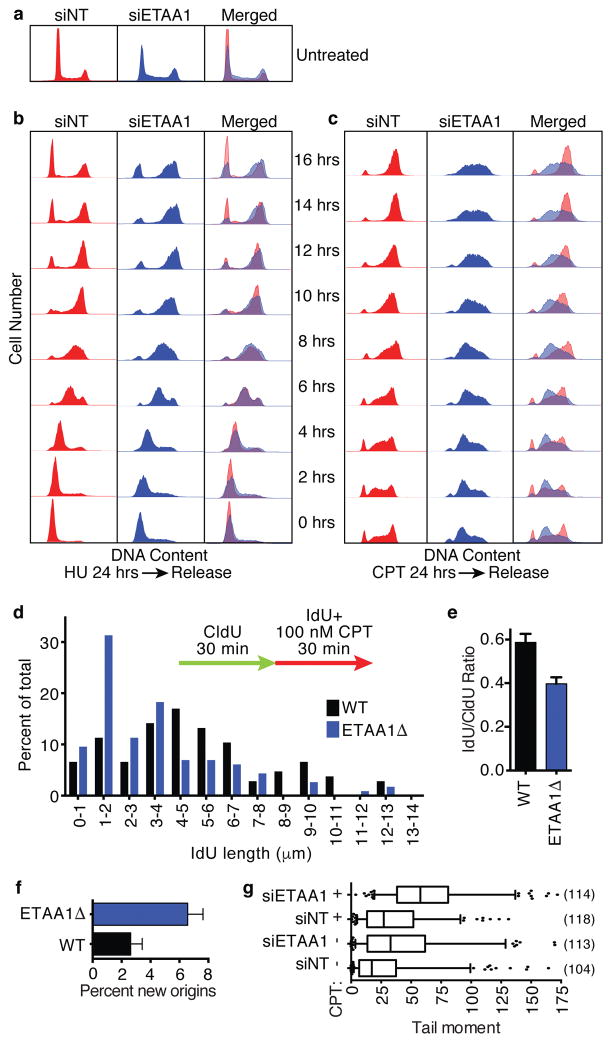
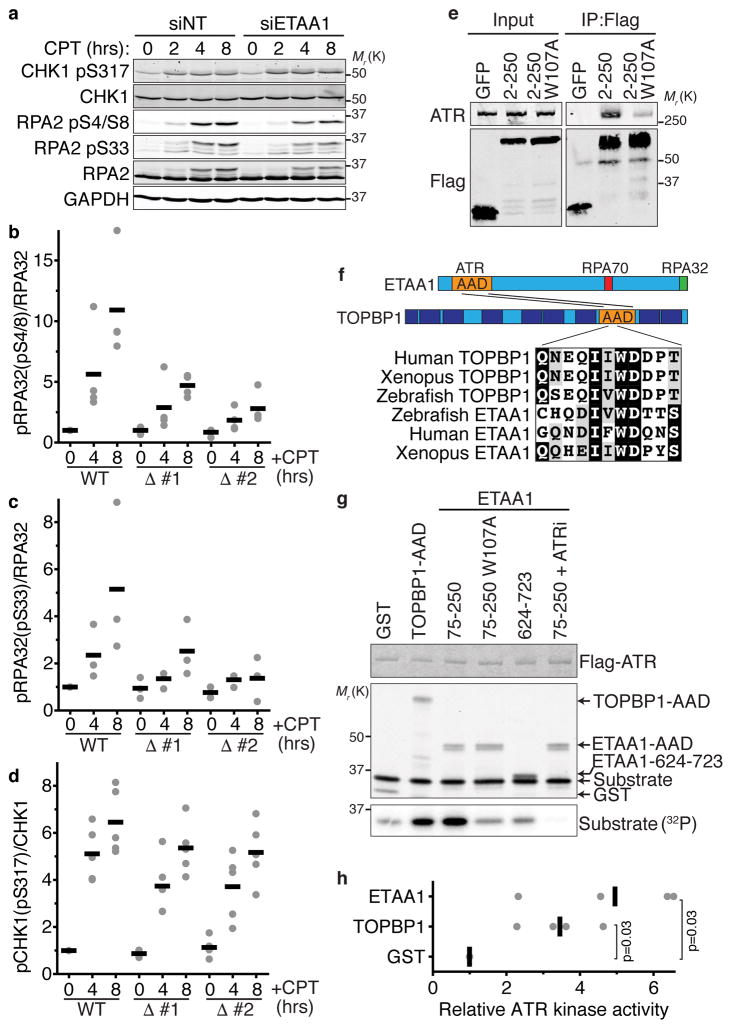
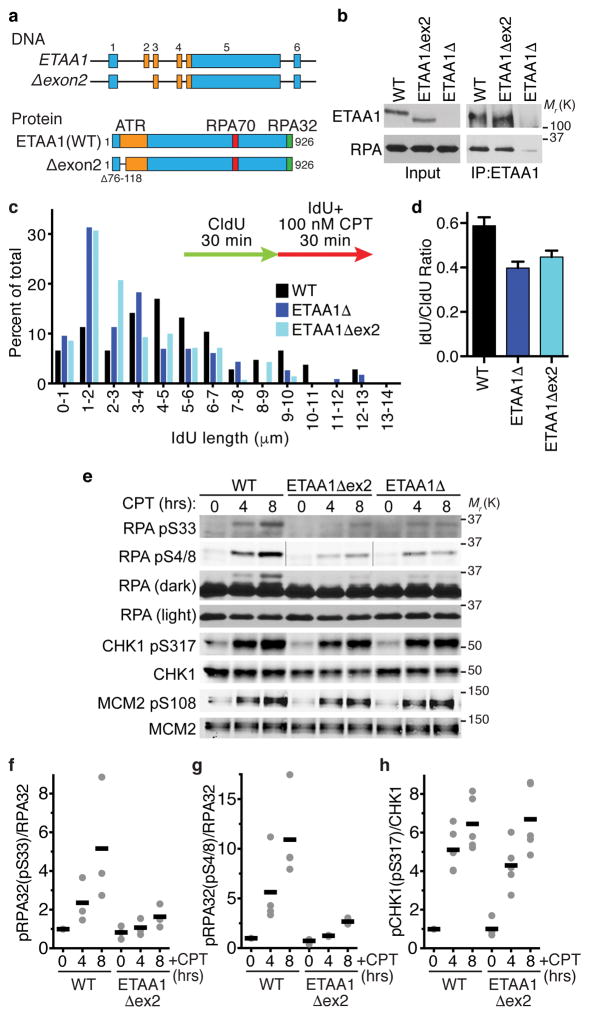
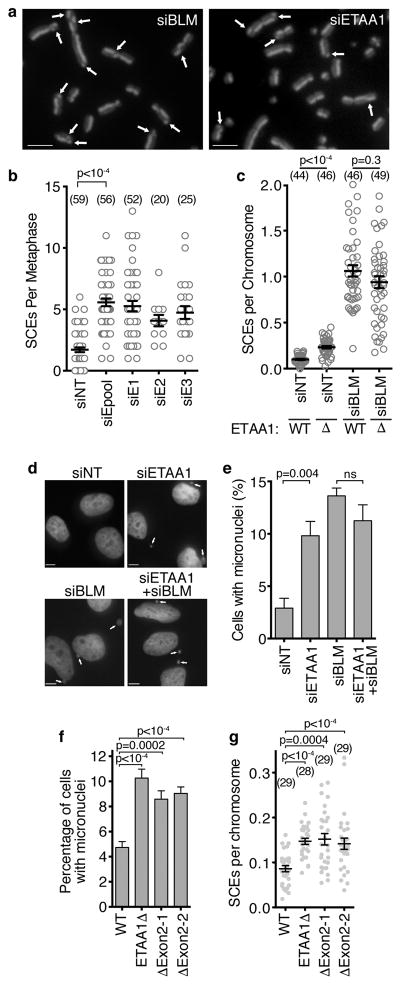
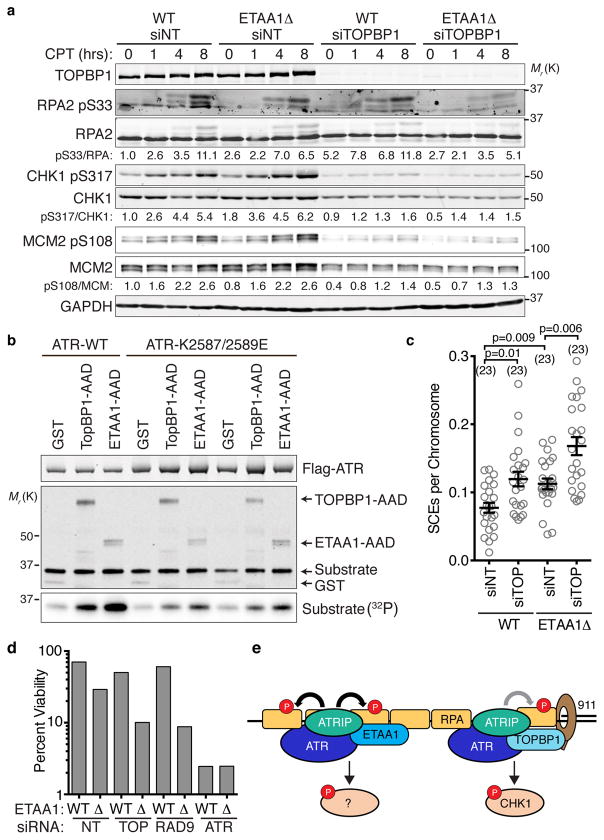
The ATR checkpoint kinase coordinates cellular responses to DNA replication stress. Budding yeast contain three activators of Mec1 (the ATR orthologue); however, only TOPBP1 is known to activate ATR in vertebrates. We identified ETAA1 as a replication stress response protein in two proteomic screens. ETAA1-deficient cells accumulate double-strand breaks, sister chromatid exchanges, and other hallmarks of genome instability. They are also hypersensitive to replication stress and have increased frequencies of replication fork collapse. ETAA1 contains two RPA-interaction motifs that localize ETAA1 to stalled replication forks. It also interacts with several DNA damage response proteins including the BLM/TOP3α/RMI1/RMI2 and ATR/ATRIP complexes. It binds ATR/ATRIP directly using a motif with sequence similarity to the TOPBP1 ATR-activation domain; and like TOPBP1, ETAA1 acts as a direct ATR activator. ETAA1 functions in parallel to the TOPBP1/RAD9/HUS1/RAD1 pathway to regulate ATR and maintain genome stability. Thus, vertebrate cells contain at least two ATR-activating proteins.
Conflict of interest statement
The authors have no competing financial interests.
Figures
References
-
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
