

Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes
- PMID: 27723724
- PMCID: PMC8903085
- DOI: 10.1038/nmicrobiol.2016.185
Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes
Abstract
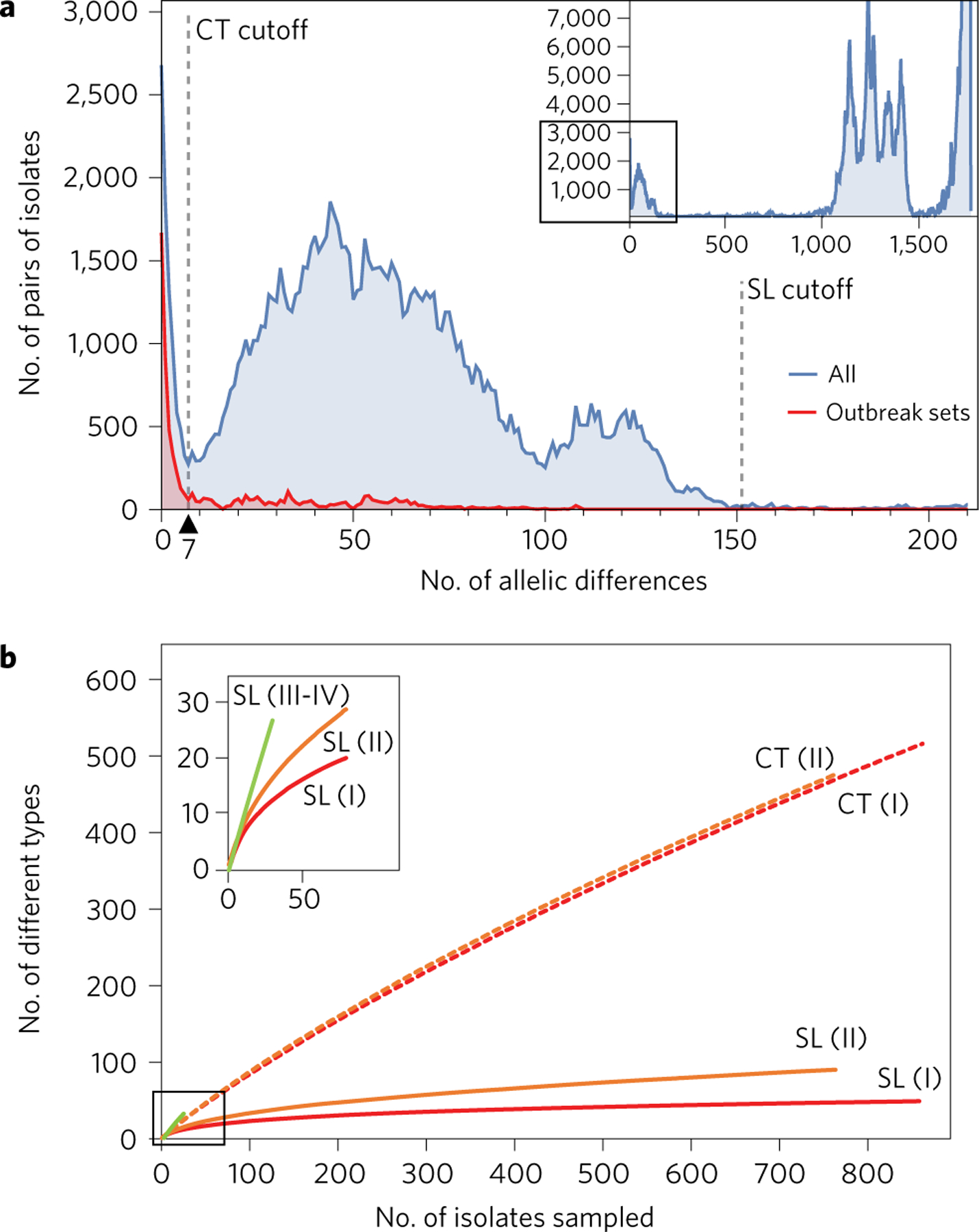
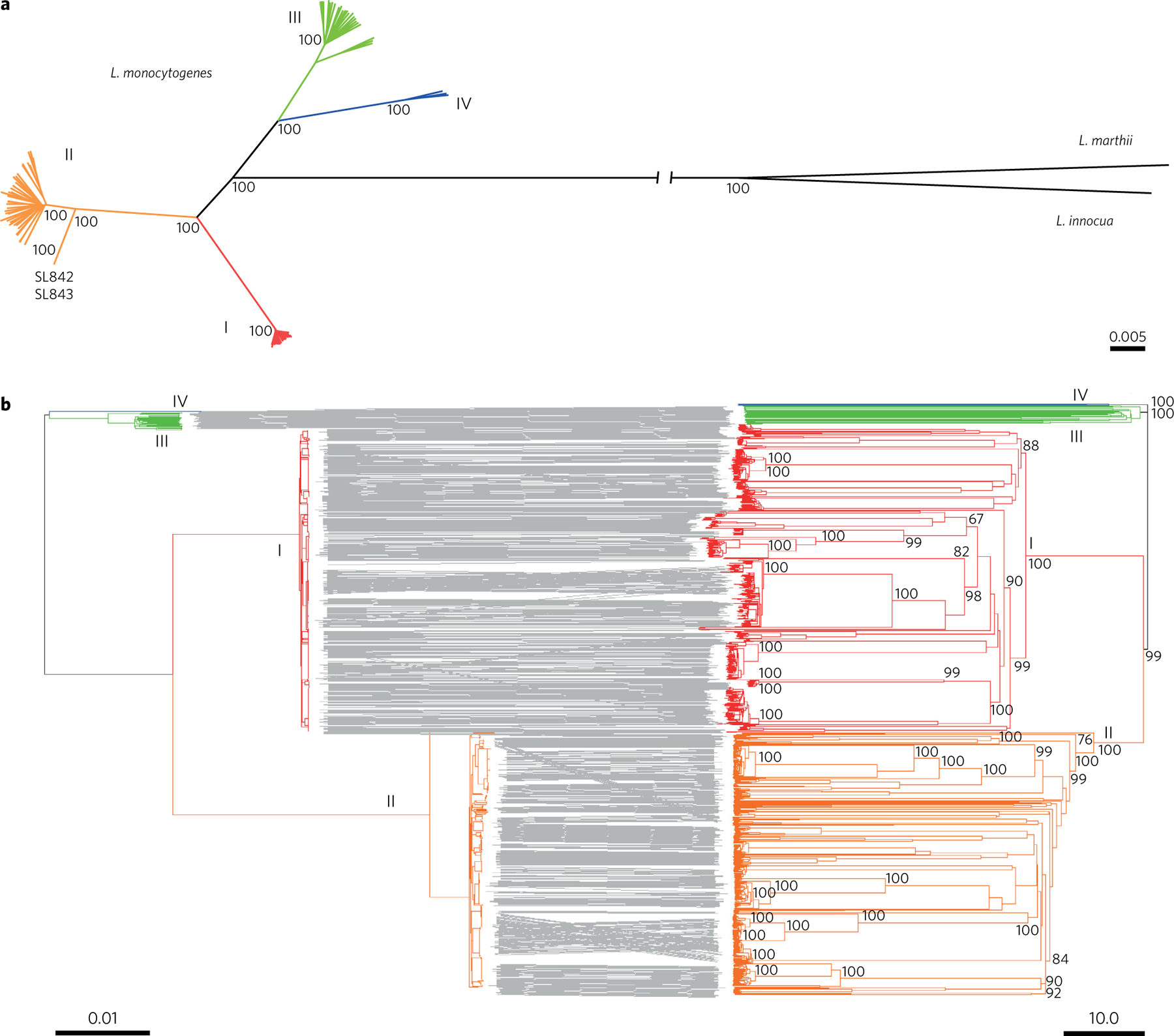
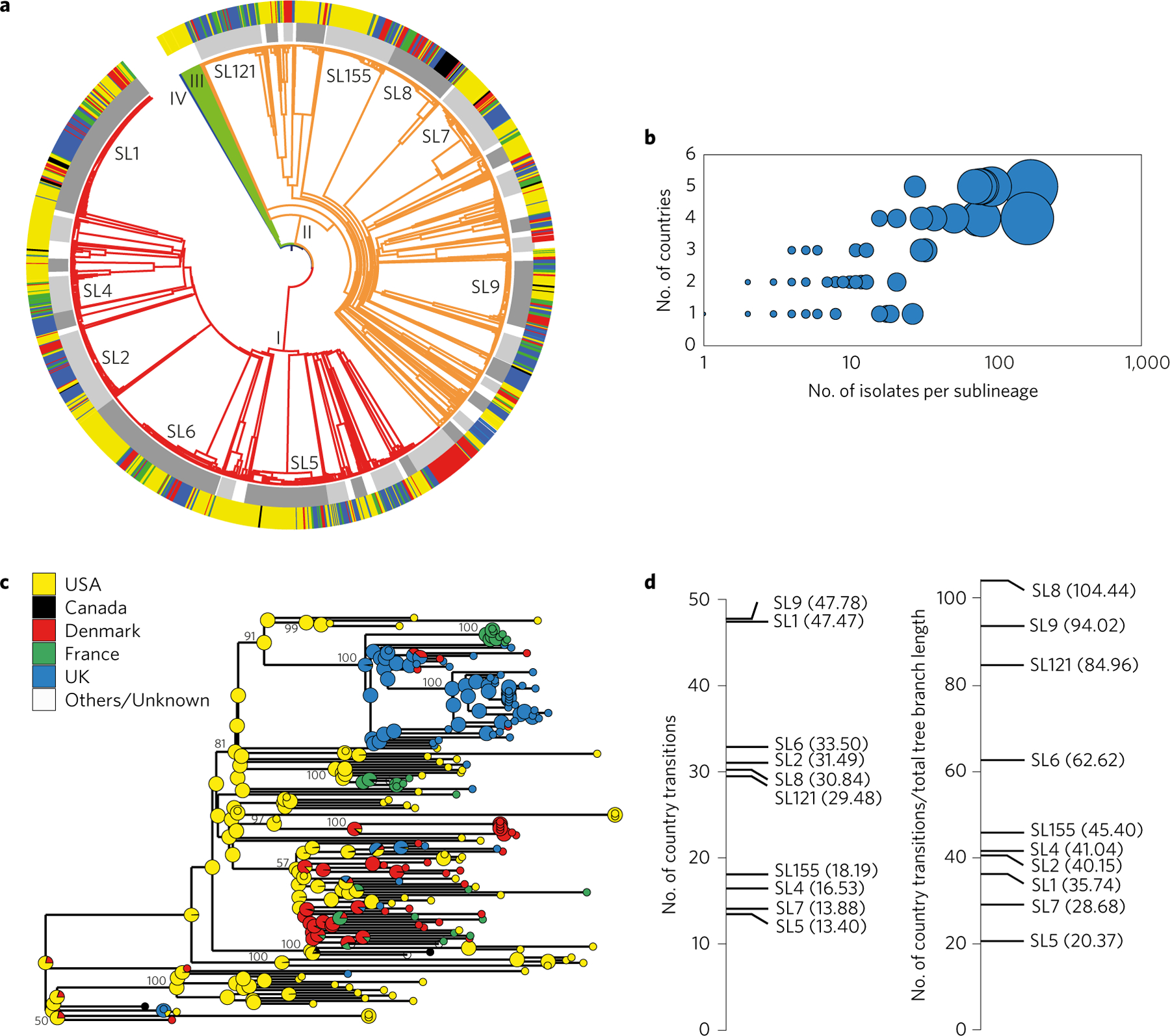
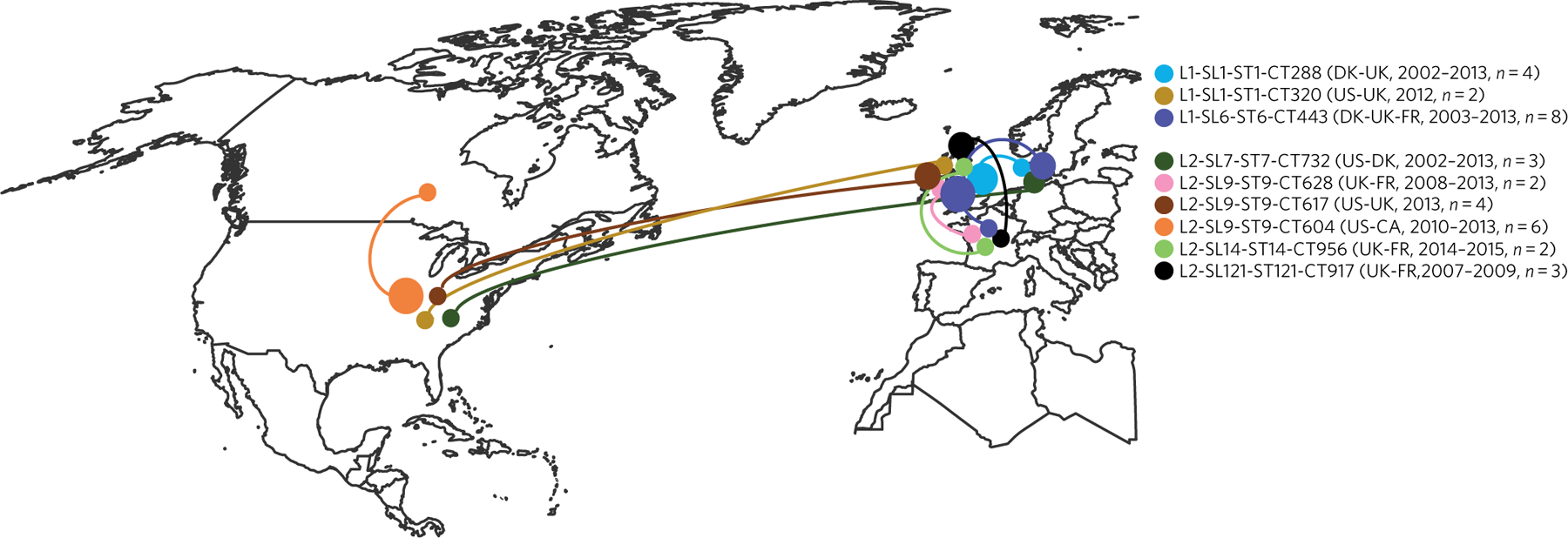
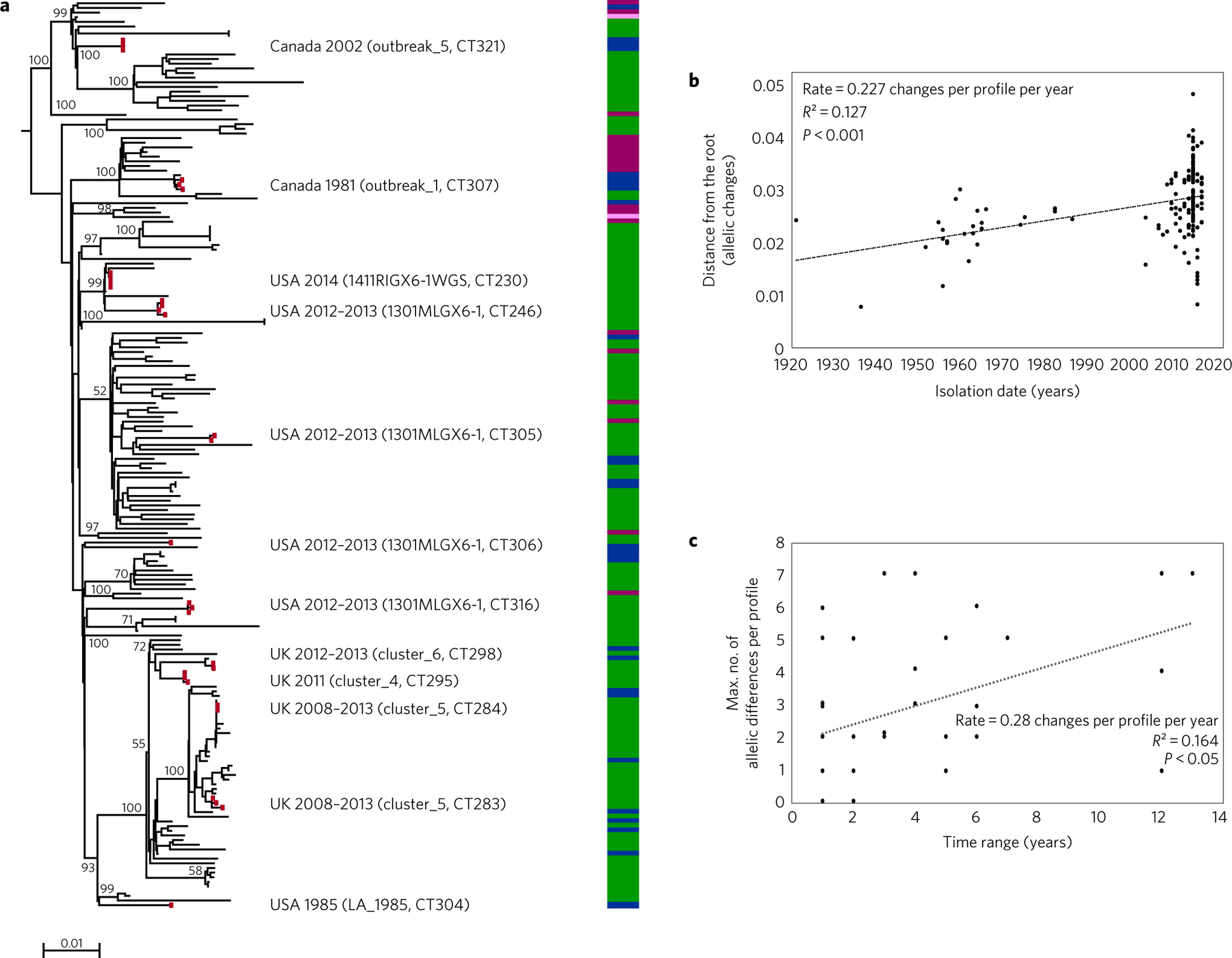
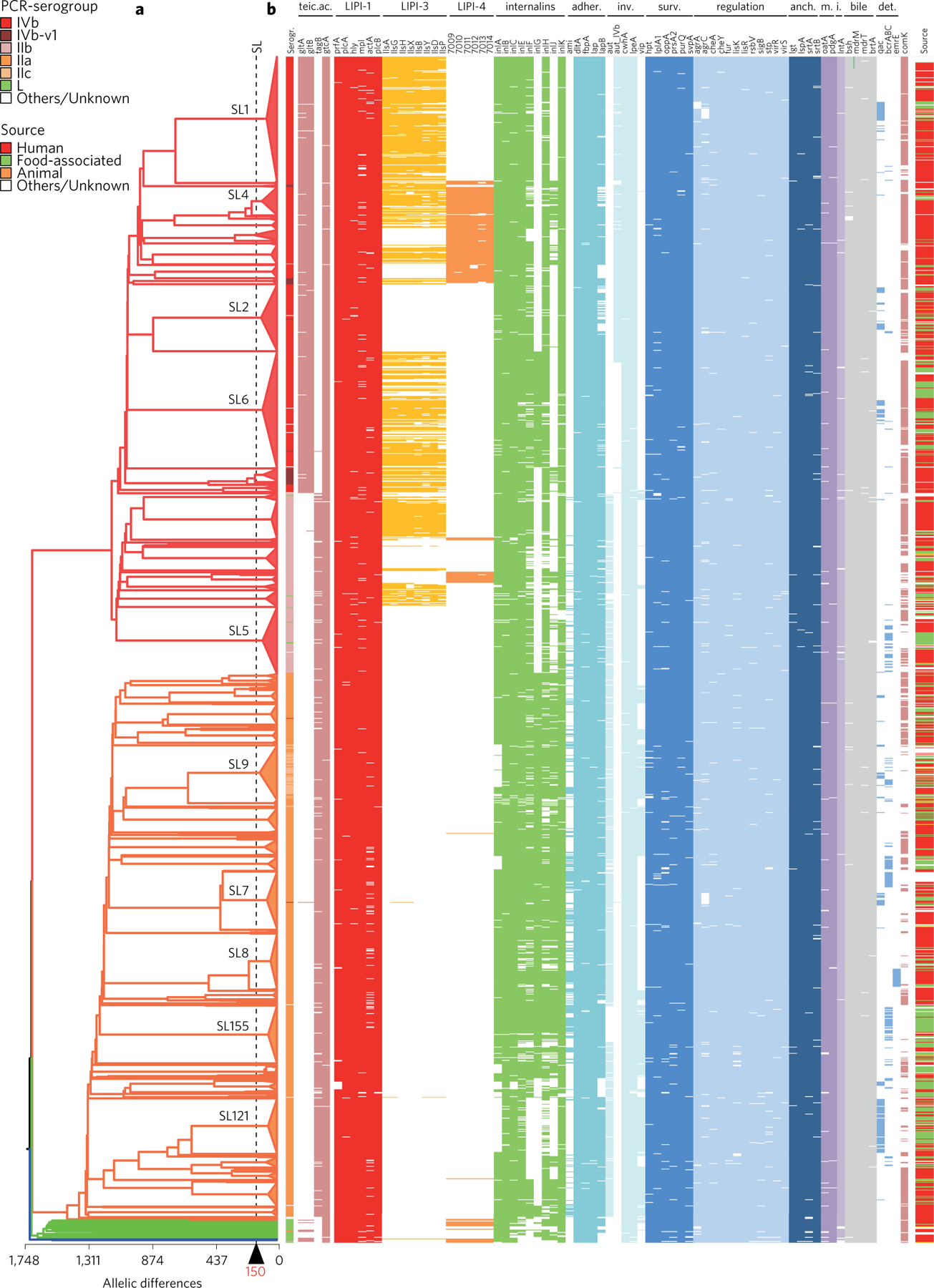
Listeria monocytogenes (Lm) is a major human foodborne pathogen. Numerous Lm outbreaks have been reported worldwide and associated with a high case fatality rate, reinforcing the need for strongly coordinated surveillance and outbreak control. We developed a universally applicable genome-wide strain genotyping approach and investigated the population diversity of Lm using 1,696 isolates from diverse sources and geographical locations. We define, with unprecedented precision, the population structure of Lm, demonstrate the occurrence of international circulation of strains and reveal the extent of heterogeneity in virulence and stress resistance genomic features among clinical and food isolates. Using historical isolates, we show that the evolutionary rate of Lm from lineage I and lineage II is low (∼2.5 × 10-7 substitutions per site per year, as inferred from the core genome) and that major sublineages (corresponding to so-called 'epidemic clones') are estimated to be at least 50-150 years old. This work demonstrates the urgent need to monitor Lm strains at the global level and provides the unified approach needed for global harmonization of Lm genome-based typing and population biology.
Conflict of interest statement
Competing interests
H.P. and B.P. are co-developers of the BioNumerics software mentioned in the manuscript. The remaining authors declare no competing interests.
Figures
References
-
- van Belkum A et al. Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Clin. Microbiol. Infect 13, 1–46 (2007). - PubMed
-
- Bogner P, Capua I, Cox NJ, Lipman DJ & Others. A global initiative on sharing avian flu data. Nature 442, 981–981 (2006).
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
