

Increased global transcription activity as a mechanism of replication stress in cancer
- PMID: 27725641
- PMCID: PMC5062618
- DOI: 10.1038/ncomms13087
Increased global transcription activity as a mechanism of replication stress in cancer
Abstract
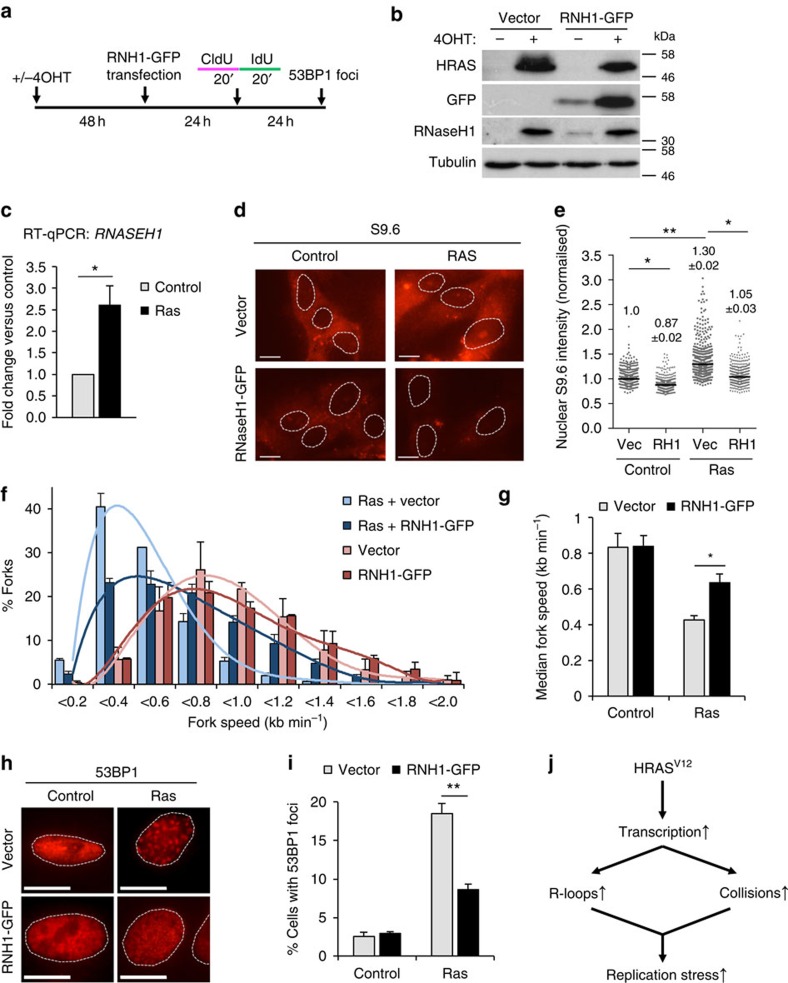
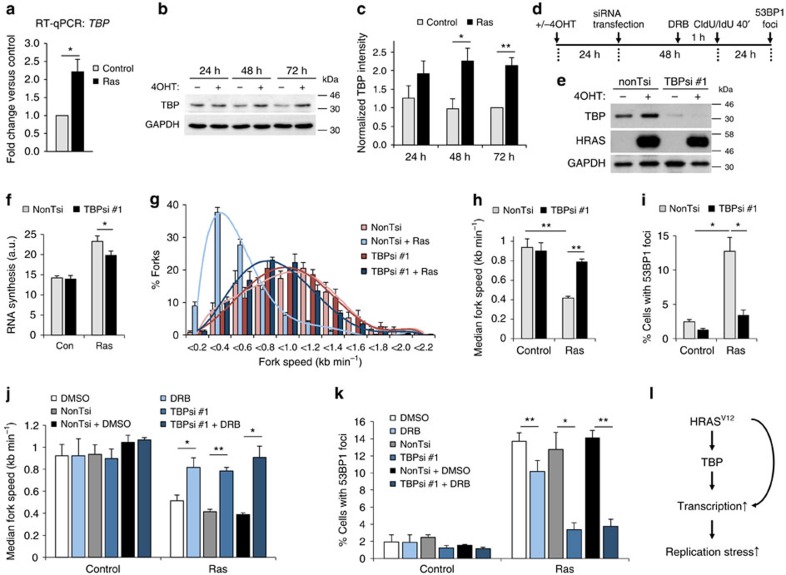
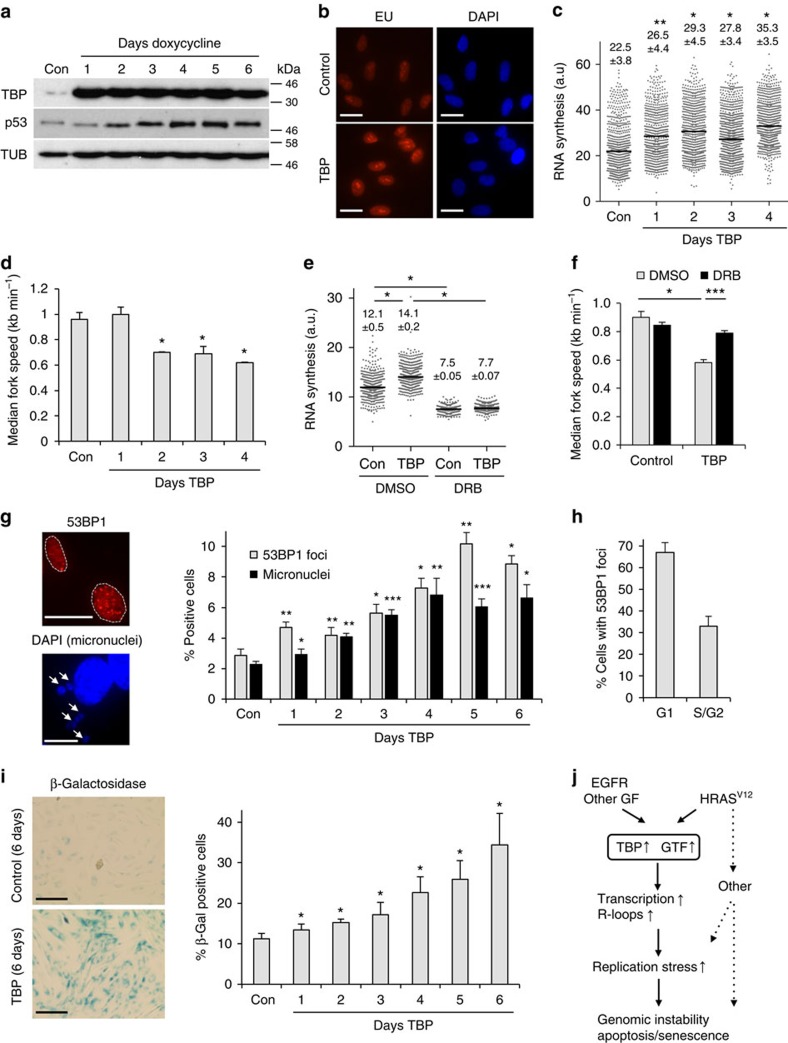
Cancer is a disease associated with genomic instability that often results from oncogene activation. This in turn leads to hyperproliferation and replication stress. However, the molecular mechanisms that underlie oncogene-induced replication stress are still poorly understood. Oncogenes such as HRASV12 promote proliferation by upregulating general transcription factors to stimulate RNA synthesis. Here we investigate whether this increase in transcription underlies oncogene-induced replication stress. We show that in cells overexpressing HRASV12, elevated expression of the general transcription factor TATA-box binding protein (TBP) leads to increased RNA synthesis, which together with R-loop accumulation results in replication fork slowing and DNA damage. Furthermore, overexpression of TBP alone causes the hallmarks of oncogene-induced replication stress, including replication fork slowing, DNA damage and senescence. Consequently, we reveal that increased transcription can be a mechanism of oncogene-induced DNA damage, providing a molecular link between upregulation of the transcription machinery and genomic instability in cancer.
Figures
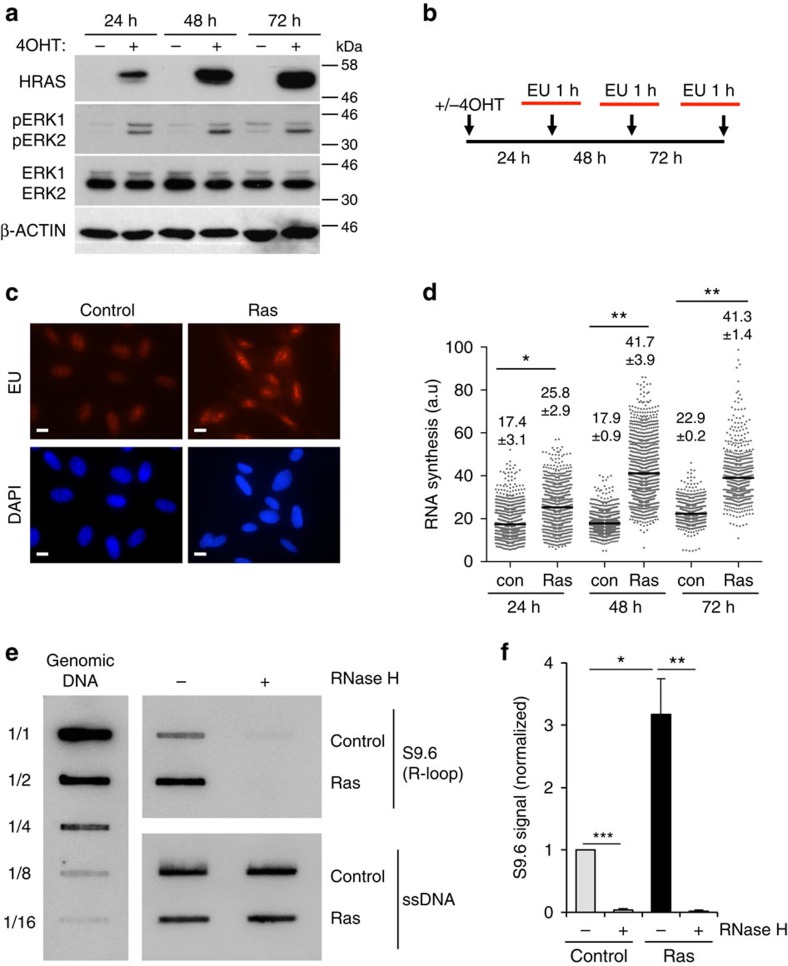
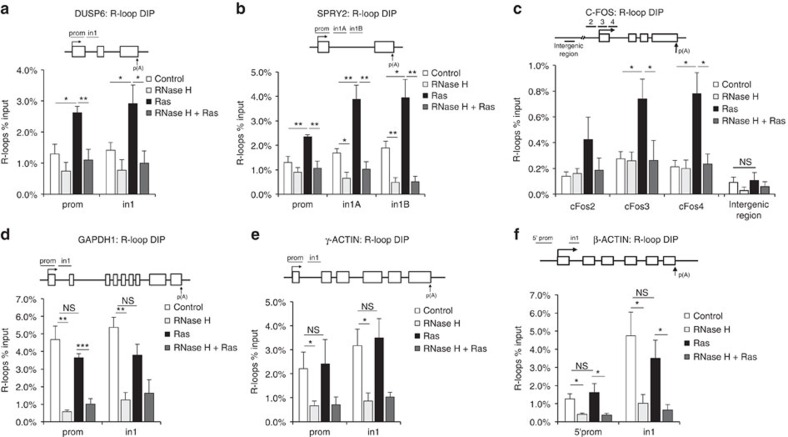
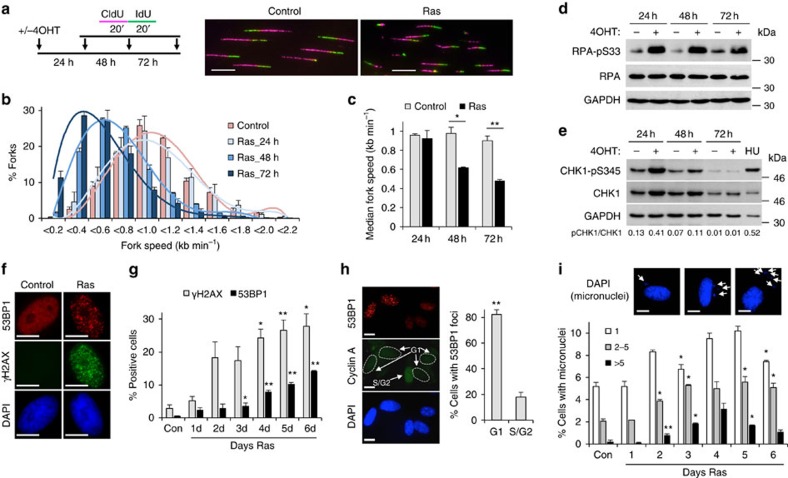
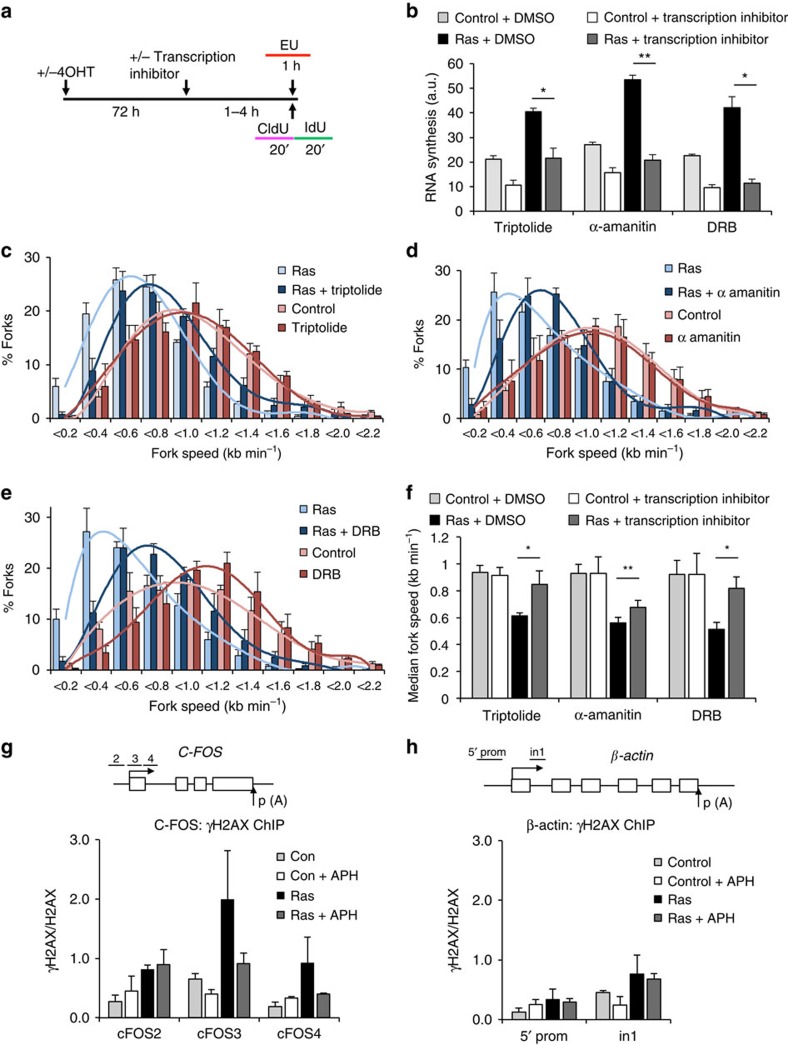
References
-
- Kotsantis P., Jones R. M., Higgs M. R. & Petermann E. Cancer therapy and replication stress: forks on the road to perdition. Adv. Clin. Chem. 69, 91–138 (2015). - PubMed
-
- Bartkova J. et al.. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 (2006). - PubMed
-
- Di Micco R. et al.. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
