

High-Throughput Identification of Adaptive Mutations in Experimentally Evolved Yeast Populations
- PMID: 27727276
- PMCID: PMC5065121
- DOI: 10.1371/journal.pgen.1006339
High-Throughput Identification of Adaptive Mutations in Experimentally Evolved Yeast Populations
Abstract
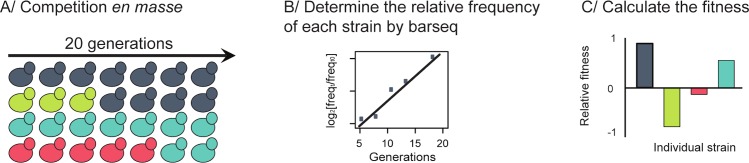
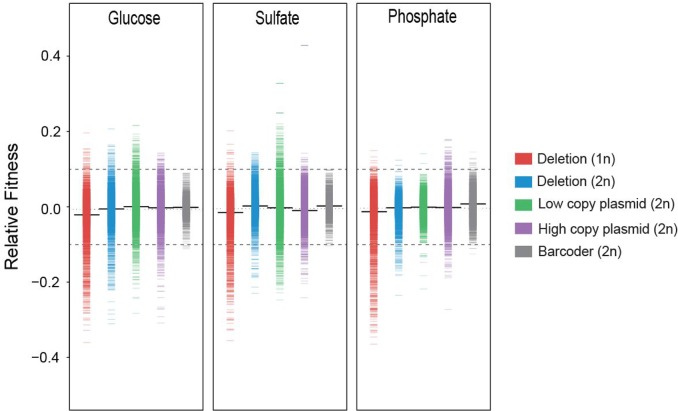
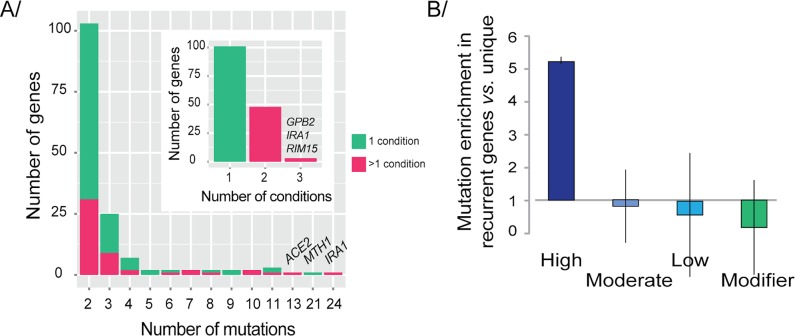
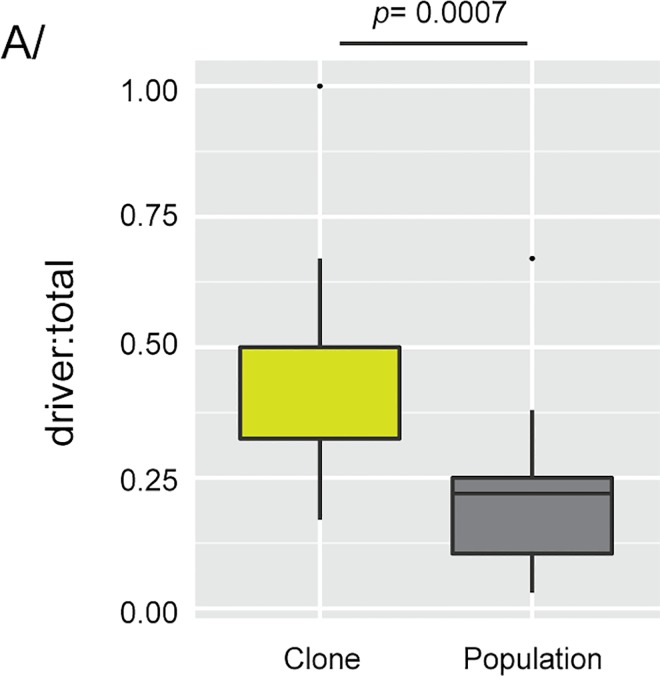
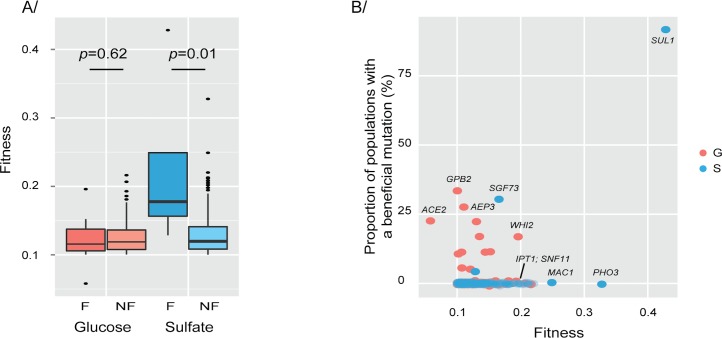
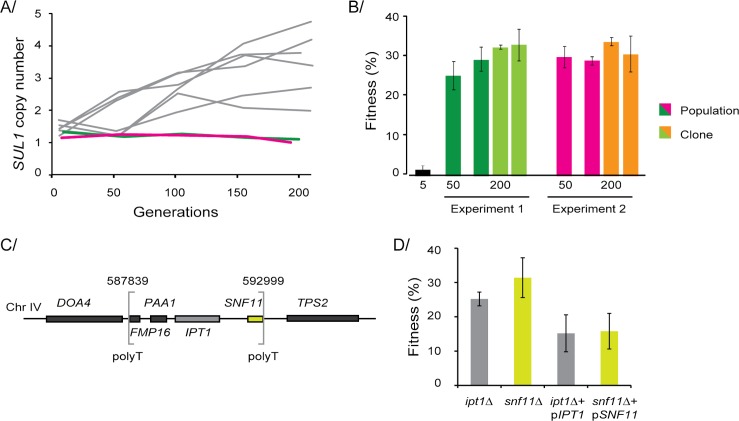
High-throughput sequencing has enabled genetic screens that can rapidly identify mutations that occur during experimental evolution. The presence of a mutation in an evolved lineage does not, however, constitute proof that the mutation is adaptive, given the well-known and widespread phenomenon of genetic hitchhiking, in which a non-adaptive or even detrimental mutation can co-occur in a genome with a beneficial mutation and the combined genotype is carried to high frequency by selection. We approximated the spectrum of possible beneficial mutations in Saccharomyces cerevisiae using sets of single-gene deletions and amplifications of almost all the genes in the S. cerevisiae genome. We determined the fitness effects of each mutation in three different nutrient-limited conditions using pooled competitions followed by barcode sequencing. Although most of the mutations were neutral or deleterious, ~500 of them increased fitness. We then compared those results to the mutations that actually occurred during experimental evolution in the same three nutrient-limited conditions. On average, ~35% of the mutations that occurred during experimental evolution were predicted by the systematic screen to be beneficial. We found that the distribution of fitness effects depended on the selective conditions. In the phosphate-limited and glucose-limited conditions, a large number of beneficial mutations of nearly equivalent, small effects drove the fitness increases. In the sulfate-limited condition, one type of mutation, the amplification of the high-affinity sulfate transporter, dominated. In the absence of that mutation, evolution in the sulfate-limited condition involved mutations in other genes that were not observed previously-but were predicted by the systematic screen. Thus, gross functional screens have the potential to predict and identify adaptive mutations that occur during experimental evolution.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
