

The Autonomic Nervous System Regulates the Heart Rate through cAMP-PKA Dependent and Independent Coupled-Clock Pacemaker Cell Mechanisms
- PMID: 27729868
- PMCID: PMC5037226
- DOI: 10.3389/fphys.2016.00419
The Autonomic Nervous System Regulates the Heart Rate through cAMP-PKA Dependent and Independent Coupled-Clock Pacemaker Cell Mechanisms
Abstract
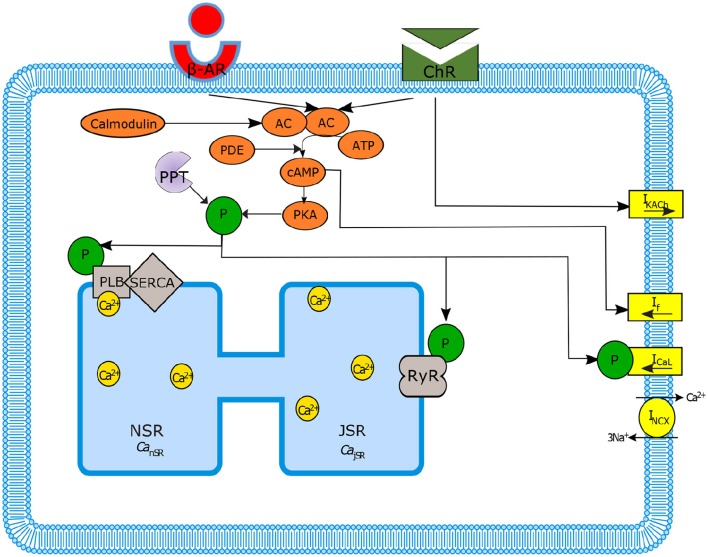
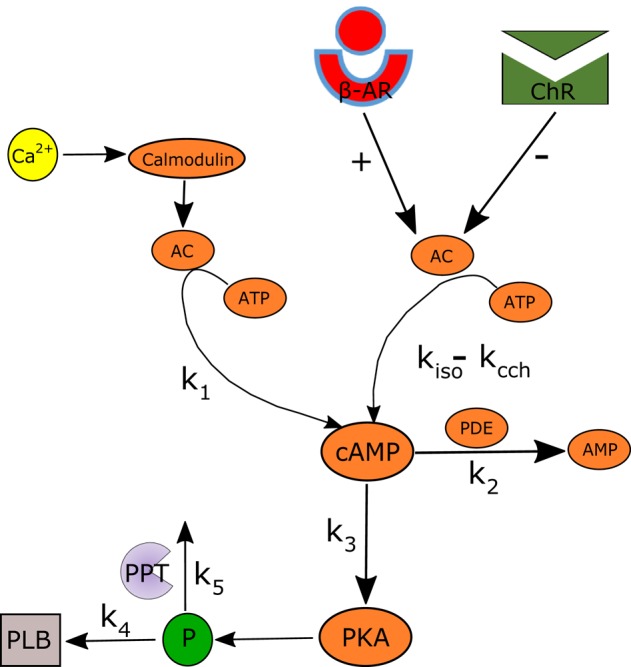
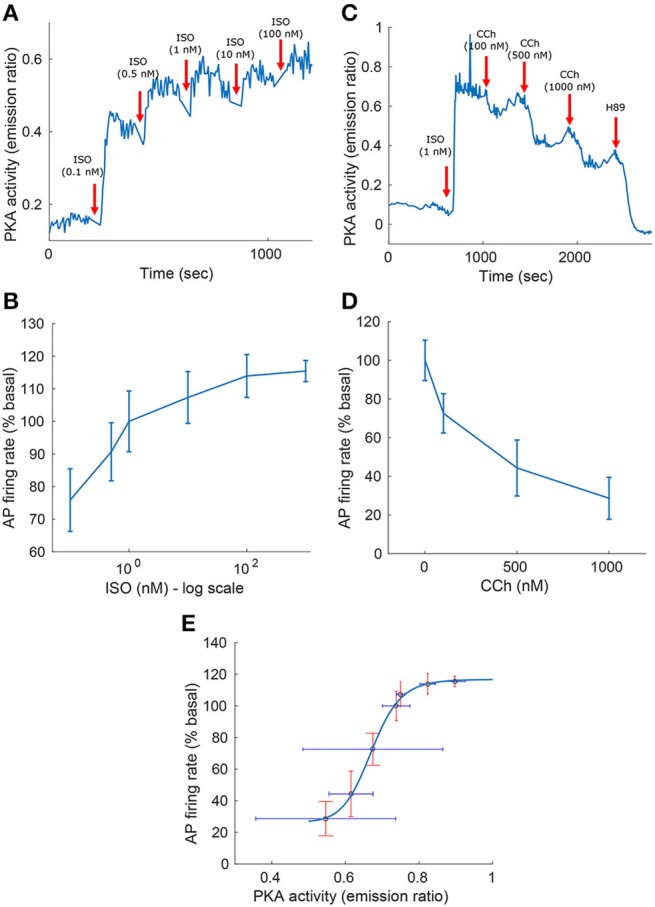
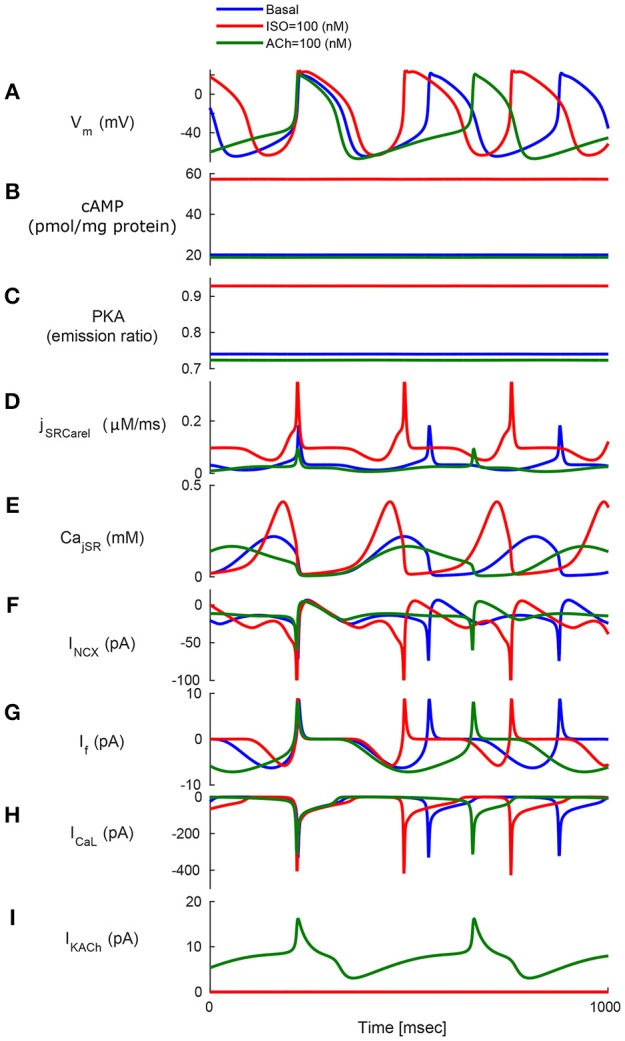
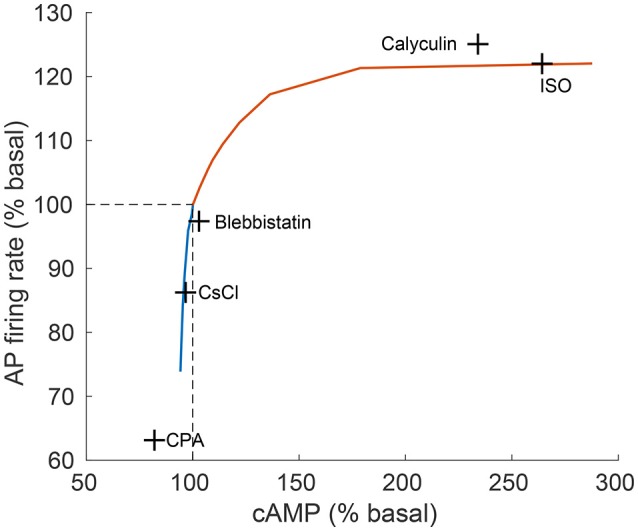
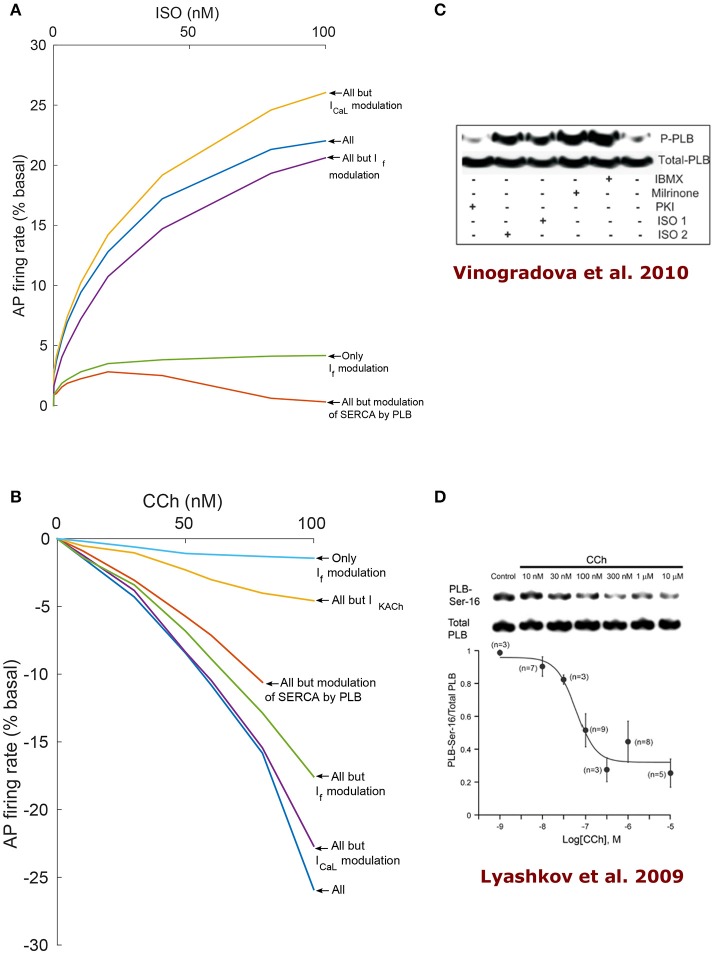
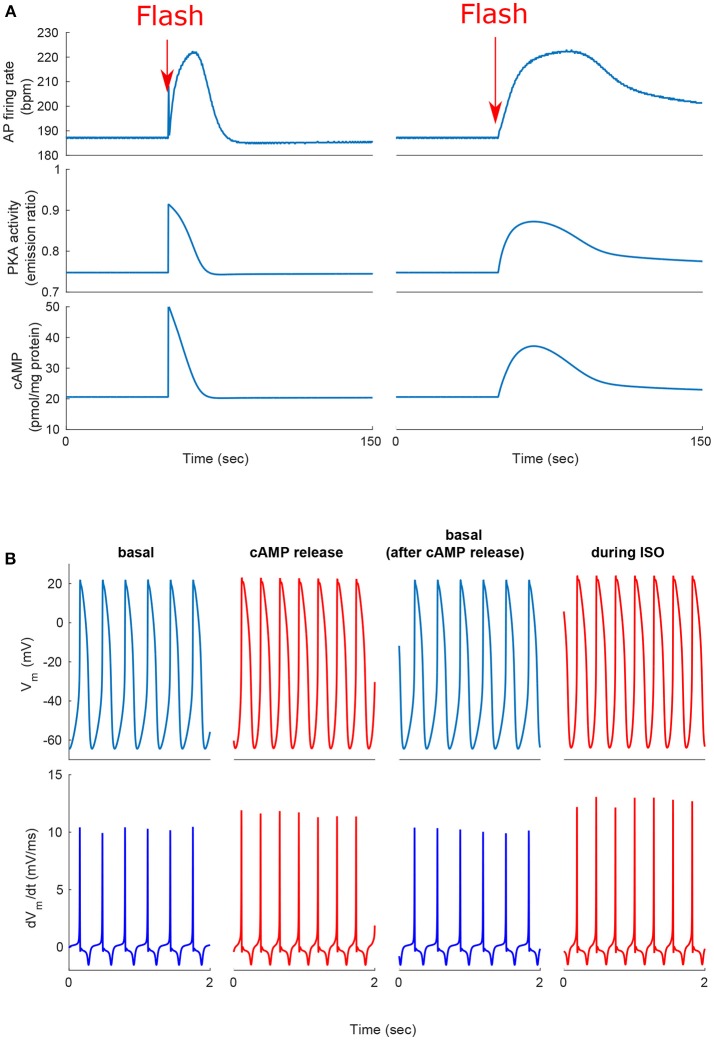
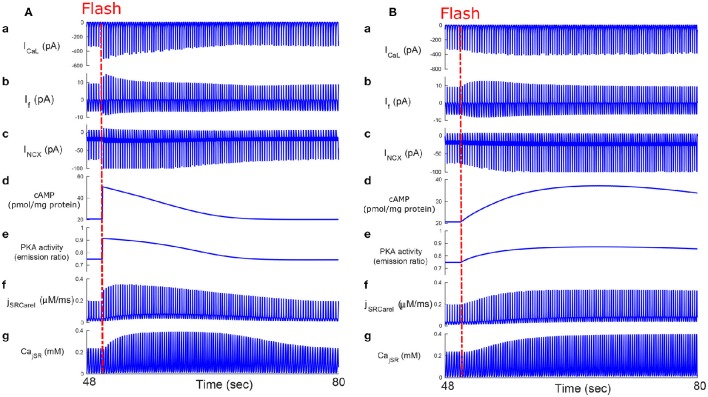
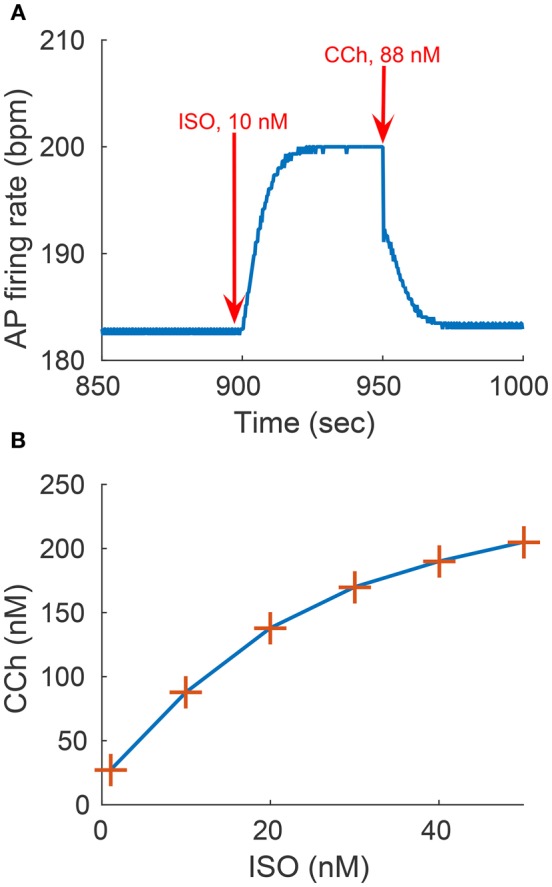
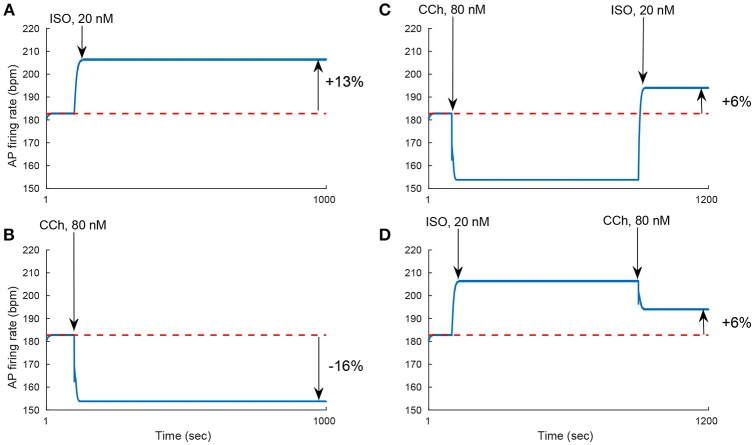
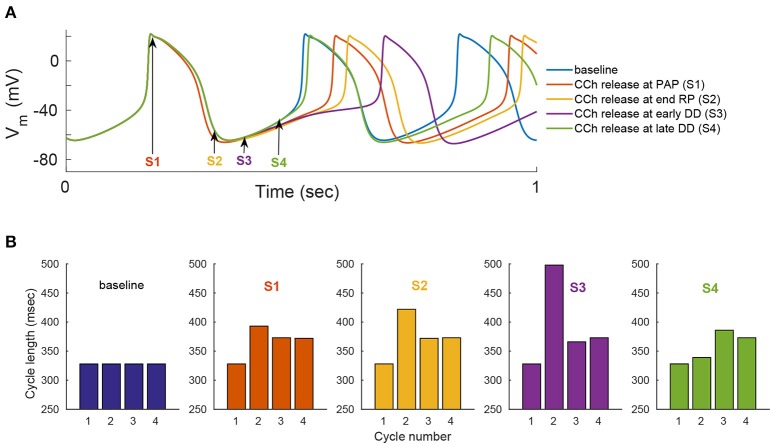
Sinoatrial nodal cells (SANCs) generate spontaneous action potentials (APs) that control the cardiac rate. The brain modulates SANC automaticity, via the autonomic nervous system, by stimulating membrane receptors that activate (adrenergic) or inactivate (cholinergic) adenylyl cyclase (AC). However, these opposing afferents are not simply additive. We showed that activation of adrenergic signaling increases AC-cAMP/PKA signaling, which mediates the increase in the SANC AP firing rate (i.e., positive chronotropic modulation). However, there is a limited understanding of the underlying internal pacemaker mechanisms involved in the crosstalk between cholinergic receptors and the decrease in the SANC AP firing rate (i.e., negative chronotropic modulation). We hypothesize that changes in AC-cAMP/PKA activity are crucial for mediating either decrease or increase in the AP firing rate and that the change in rate is due to both internal and membrane mechanisms. In cultured adult rabbit pacemaker cells infected with an adenovirus expressing the FRET sensor AKAR3, PKA activity and AP firing rate were tightly linked in response to either adrenergic receptor stimulation (by isoproterenol, ISO) or cholinergic stimulation (by carbachol, CCh). To identify the main molecular targets that mediate between PKA signaling and pacemaker function, we developed a mechanistic computational model. The model includes a description of autonomic-nervous receptors, post- translation signaling cascades, membrane molecules, and internal pacemaker mechanisms. Yielding results similar to those of the experiments, the model simulations faithfully reproduce the changes in AP firing rate in response to CCh or ISO or a combination of both (i.e., accentuated antagonism). Eliminating AC-cAMP-PKA signaling abolished the core effect of autonomic receptor stimulation on the AP firing rate. Specifically, disabling the phospholamban modulation of the SERCA activity resulted in a significantly reduced effect of CCh and a failure to increase the AP firing rate under ISO stimulation. Directly activating internal pacemaker mechanisms led to a similar extent of changes in the AP firing rate with respect to brain receptor stimulation. Thus, Ca2+ and cAMP/PKA-dependent phosphorylation limits the rate and magnitude of chronotropic changes in the spontaneous AP firing rate.
Keywords: autonomic regulation; cAMP-PKA signaling; coupled clock system; mathematical model; pacemaker cells; phospholamban.
Figures
References
-
- Baruscotti M., Bucchi A., Milanesi R., Paina M., Barbuti A., Gnecchi-Ruscone T., et al. . (2015). A gain-of-function mutation in the cardiac pacemaker HCN4 channel increasing cAMP sensitivity is associated with familial Inappropriate Sinus Tachycardia. Eur. Heart J.. [Epub ahead of print]. 10.1093/eurheartj/ehv582 - DOI - PubMed
-
- Bogdanov K. Y., Maltsev V. A., Vinogradova T. M., Lyashkov A. E., Spurgeon H. A., Stern M. D., et al. . (2006). Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ. Res. 99, 979–987. 10.1161/01.RES.0000247933.66532.0b - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous