

Antibody-drug conjugates: recent advances in conjugation and linker chemistries
- PMID: 27743348
- PMCID: PMC5777969
- DOI: 10.1007/s13238-016-0323-0
Antibody-drug conjugates: recent advances in conjugation and linker chemistries
Abstract
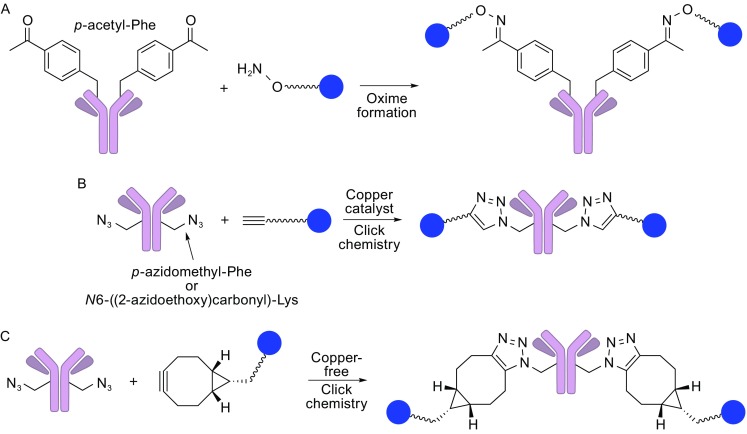
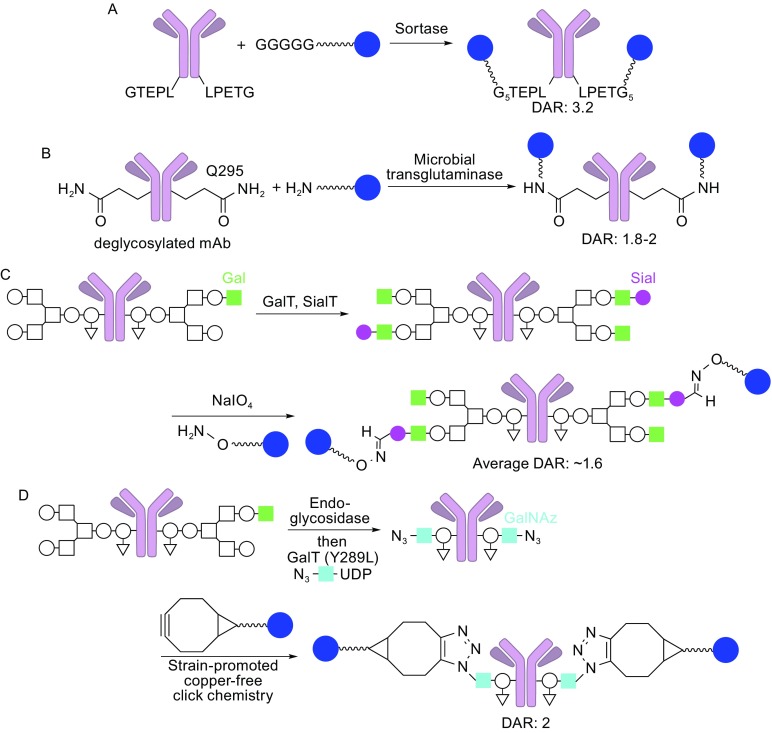
The antibody-drug conjugate (ADC), a humanized or human monoclonal antibody conjugated with highly cytotoxic small molecules (payloads) through chemical linkers, is a novel therapeutic format and has great potential to make a paradigm shift in cancer chemotherapy. This new antibody-based molecular platform enables selective delivery of a potent cytotoxic payload to target cancer cells, resulting in improved efficacy, reduced systemic toxicity, and preferable pharmacokinetics (PK)/pharmacodynamics (PD) and biodistribution compared to traditional chemotherapy. Boosted by the successes of FDA-approved Adcetris® and Kadcyla®, this drug class has been rapidly growing along with about 60 ADCs currently in clinical trials. In this article, we briefly review molecular aspects of each component (the antibody, payload, and linker) of ADCs, and then mainly discuss traditional and new technologies of the conjugation and linker chemistries for successful construction of clinically effective ADCs. Current efforts in the conjugation and linker chemistries will provide greater insights into molecular design and strategies for clinically effective ADCs from medicinal chemistry and pharmacology standpoints. The development of site-specific conjugation methodologies for constructing homogeneous ADCs is an especially promising path to improving ADC design, which will open the way for novel cancer therapeutics.
Keywords: antibody-drug conjugates; cancer; chemotherapy; conjugation; linker; site-specific conjugation.
Figures






References
-
- Behrens CR, Ha EH, Chinn LL, Bowers S, Probst G, Fitch-Bruhns M, Monteon J, Valdiosera A, Bermudez A, Liao-Chan S, et al. Antibody-drug conjugates (ADCs) derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. Mol Pharm. 2015;12:3986–3998. doi: 10.1021/acs.molpharmaceut.5b00432. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
