

The impact of recent population history on the deleterious mutation load in humans and close evolutionary relatives
- PMID: 27744216
- PMCID: PMC5161708
- DOI: 10.1016/j.gde.2016.09.006
The impact of recent population history on the deleterious mutation load in humans and close evolutionary relatives
Abstract
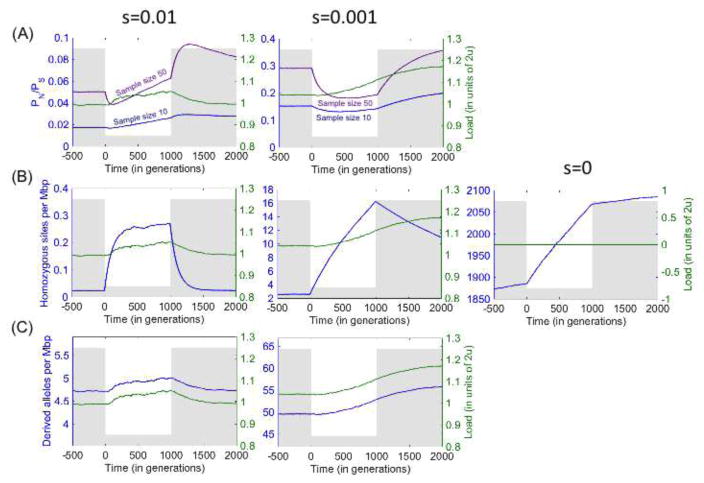
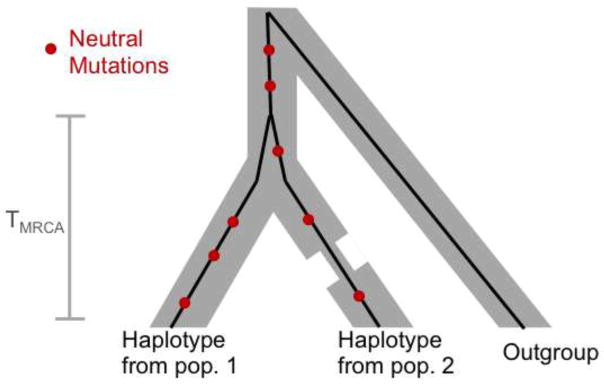
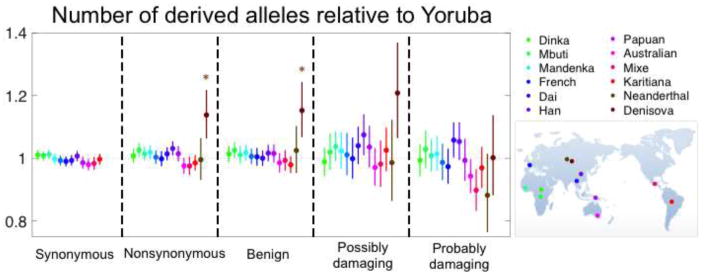
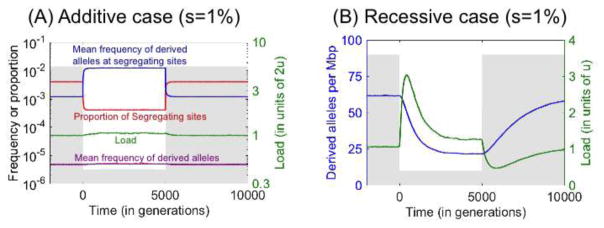
Over the past decade, there has been both great interest and confusion about whether recent demographic events-notably the Out-of-Africa-bottleneck and recent population growth-have led to differences in mutation load among human populations. The confusion can be traced to the use of different summary statistics to measure load, which lead to apparently conflicting results. We argue, however, that when statistics more directly related to load are used, the results of different studies and data sets consistently reveal little or no difference in the load of non-synonymous mutations among human populations. Theory helps to understand why no such differences are seen, as well as to predict in what settings they are to be expected. In particular, as predicted by modeling, there is evidence for changes in the load of recessive loss of function mutations in founder and inbred human populations. Also as predicted, eastern subspecies of gorilla, Neanderthals and Denisovans, who are thought to have undergone reductions in population sizes that exceed the human Out-of-Africa bottleneck in duration and severity, show evidence for increased load of non-synonymous mutations (relative to western subspecies of gorillas and modern humans, respectively). A coherent picture is thus starting to emerge about the effects of demographic history on the mutation load in populations of humans and close evolutionary relatives.
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
