

Periodontal Ehlers-Danlos Syndrome Is Caused by Mutations in C1R and C1S, which Encode Subcomponents C1r and C1s of Complement
- PMID: 27745832
- PMCID: PMC5097948
- DOI: 10.1016/j.ajhg.2016.08.019
Periodontal Ehlers-Danlos Syndrome Is Caused by Mutations in C1R and C1S, which Encode Subcomponents C1r and C1s of Complement
Abstract
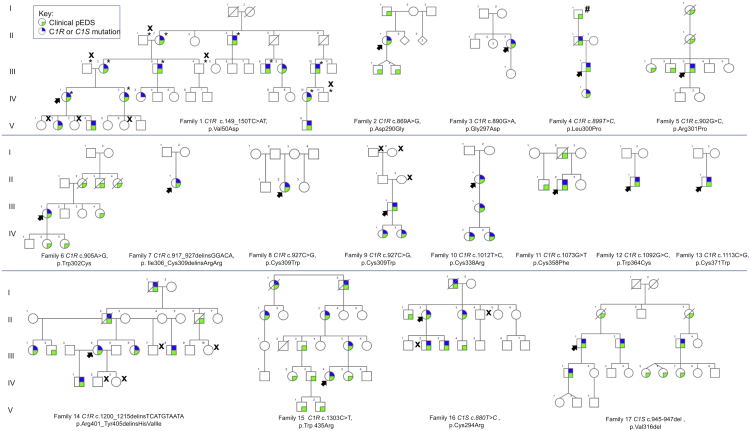
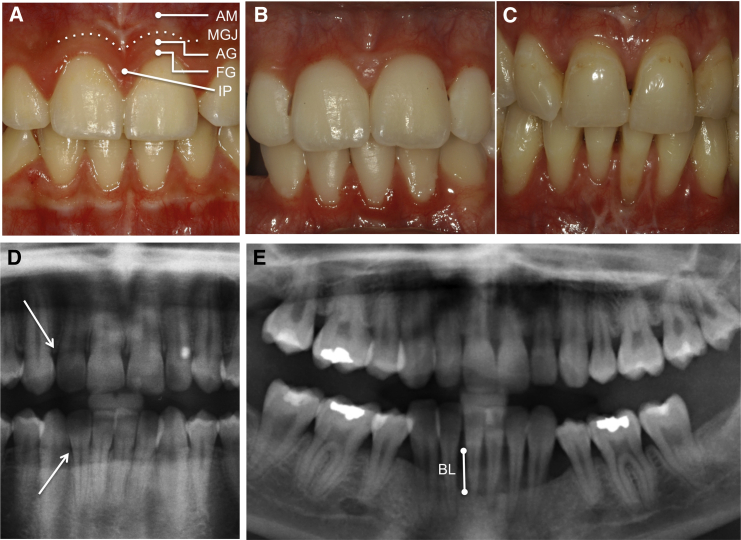
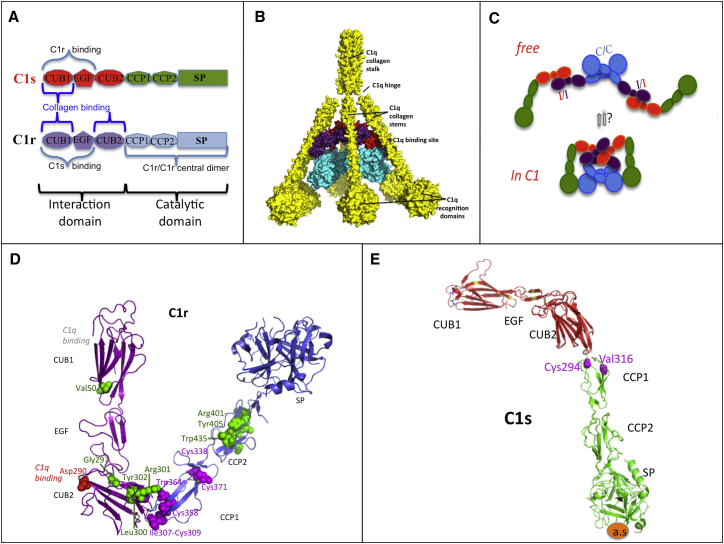
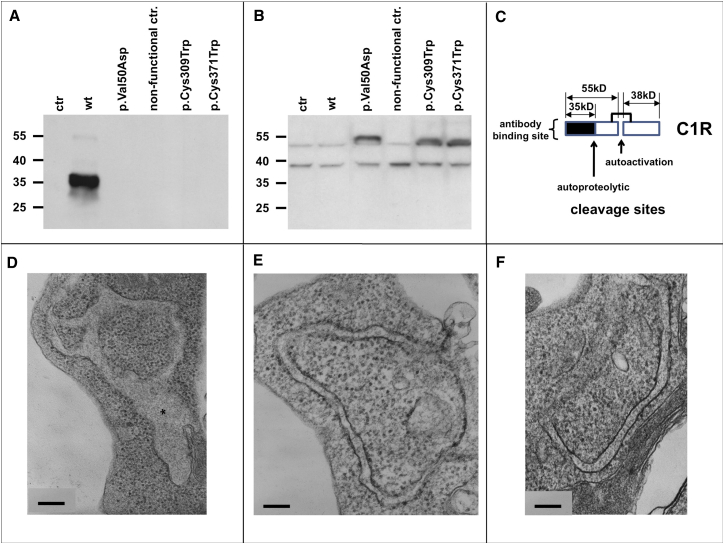
Periodontal Ehlers-Danlos syndrome (pEDS) is an autosomal-dominant disorder characterized by early-onset periodontitis leading to premature loss of teeth, joint hypermobility, and mild skin findings. A locus was mapped to an approximately 5.8 Mb region at 12p13.1 but no candidate gene was identified. In an international consortium we recruited 19 independent families comprising 107 individuals with pEDS to identify the locus, characterize the clinical details in those with defined genetic causes, and try to understand the physiological basis of the condition. In 17 of these families, we identified heterozygous missense or in-frame insertion/deletion mutations in C1R (15 families) or C1S (2 families), contiguous genes in the mapped locus that encode subunits C1r and C1s of the first component of the classical complement pathway. These two proteins form a heterotetramer that then combines with six C1q subunits. Pathogenic variants involve the subunit interfaces or inter-domain hinges of C1r and C1s and are associated with intracellular retention and mild endoplasmic reticulum enlargement. Clinical features of affected individuals in these families include rapidly progressing periodontitis with onset in the teens or childhood, a previously unrecognized lack of attached gingiva, pretibial hyperpigmentation, skin and vascular fragility, easy bruising, and variable musculoskeletal symptoms. Our findings open a connection between the inflammatory classical complement pathway and connective tissue homeostasis.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Vanakker O., Callewaert B., Malfait F., Coucke P. The genetics of soft connective tissue disorders. Annu. Rev. Genomics Hum. Genet. 2015;16:229–255. - PubMed
-
- Stewart R.E., Hollister D.W., Rimoin D.L. A new variant of Ehlers-Danlos syndrome: an autosomal dominant disorder of fragile skin, abnormal scarring, and generalized periodontitis. Birth Defects Orig. Artic. Ser. 1977;13(3B):85–93. - PubMed
-
- Rahman N., Dunstan M., Teare M.D., Hanks S., Douglas J., Coleman K., Bottomly W.E., Campbell M.E., Berglund B., Nordenskjöld M. Ehlers-Danlos syndrome with severe early-onset periodontal disease (EDS-VIII) is a distinct, heterogeneous disorder with one predisposition gene at chromosome 12p13. Am. J. Hum. Genet. 2003;73:198–204. - PMC - PubMed
-
- Reinstein E., Wang R.Y., Zhan L., Rimoin D.L., Wilcox W.R. Ehlers-Danlos type VIII, periodontitis-type: further delineation of the syndrome in a four-generation pedigree. Am. J. Med. Genet. A. 2011;155A:742–747. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
