

Variants in the Oxidoreductase PYROXD1 Cause Early-Onset Myopathy with Internalized Nuclei and Myofibrillar Disorganization
- PMID: 27745833
- PMCID: PMC5097943
- DOI: 10.1016/j.ajhg.2016.09.005
Variants in the Oxidoreductase PYROXD1 Cause Early-Onset Myopathy with Internalized Nuclei and Myofibrillar Disorganization
Abstract
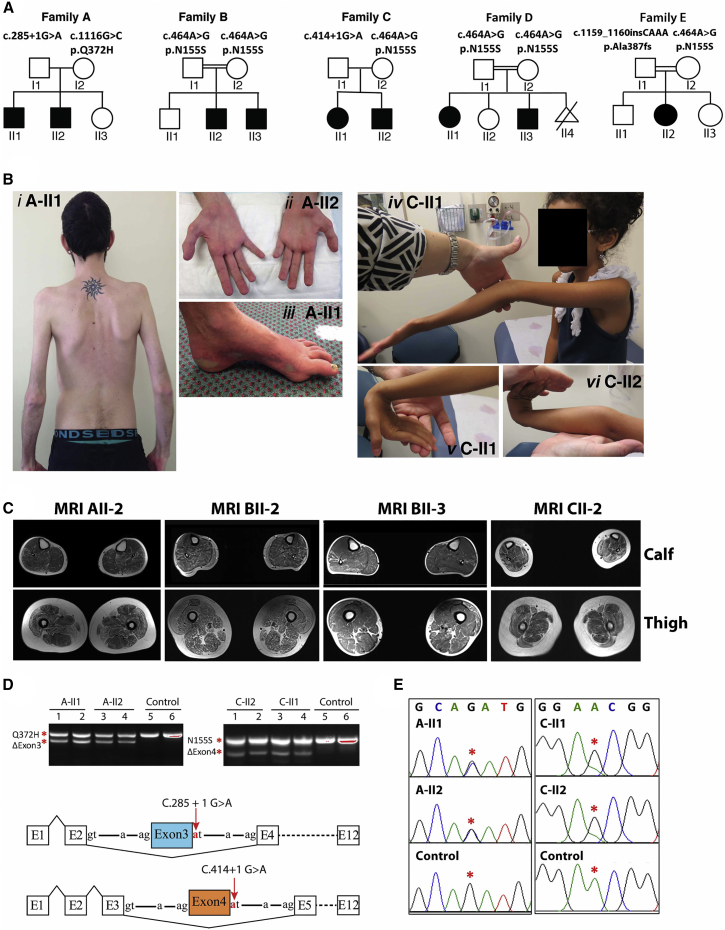
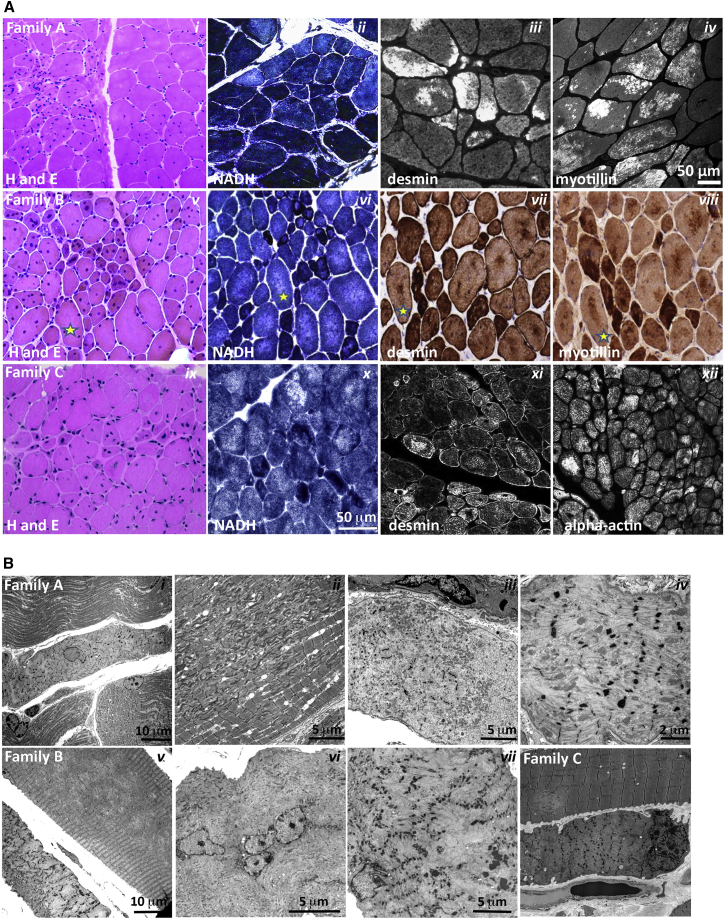
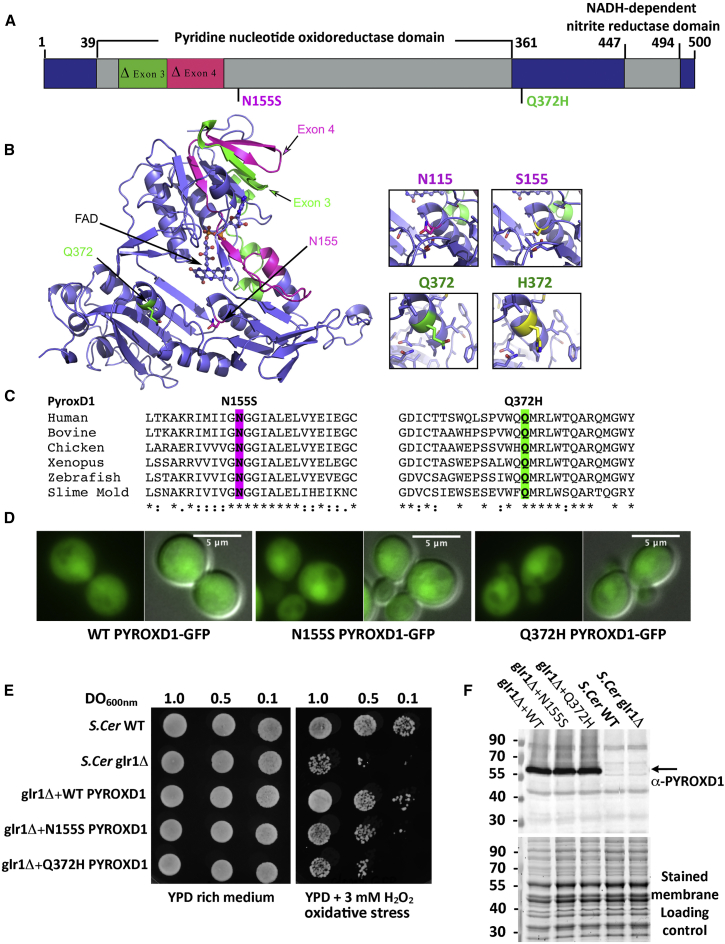
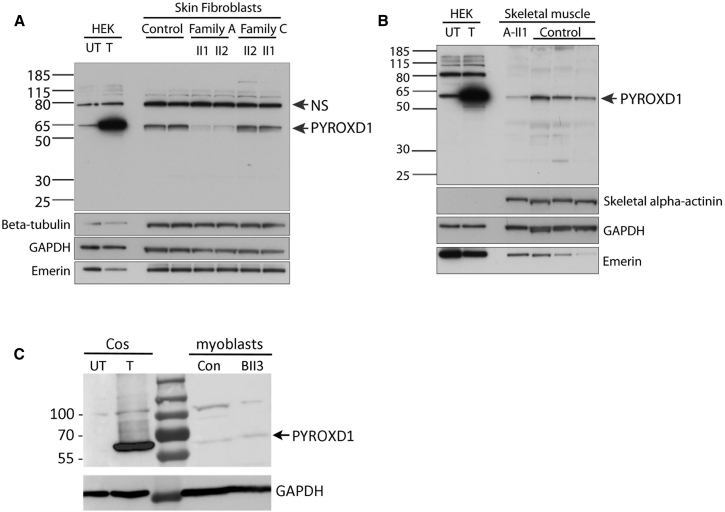
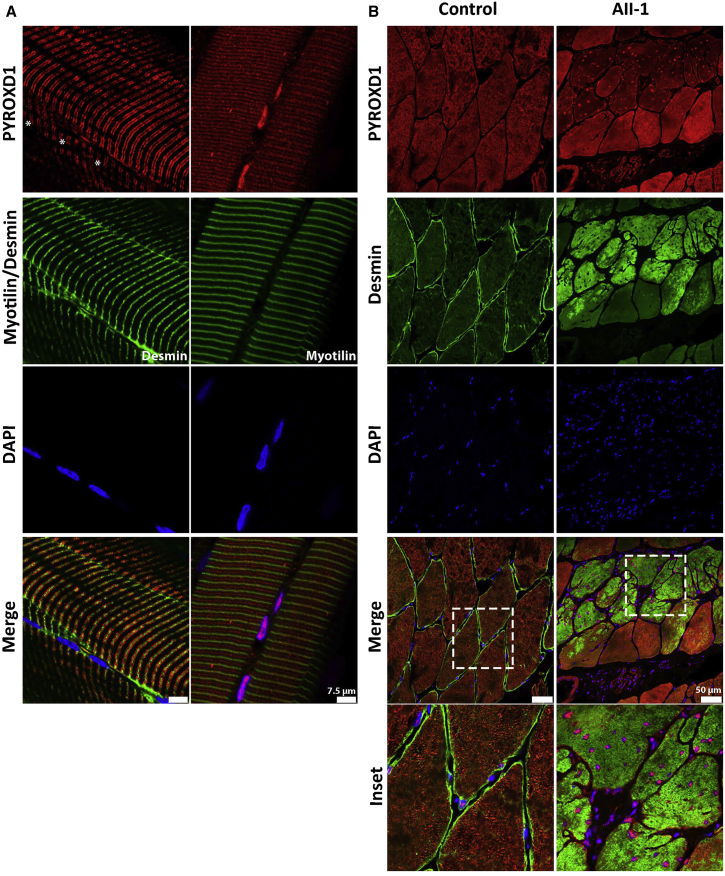
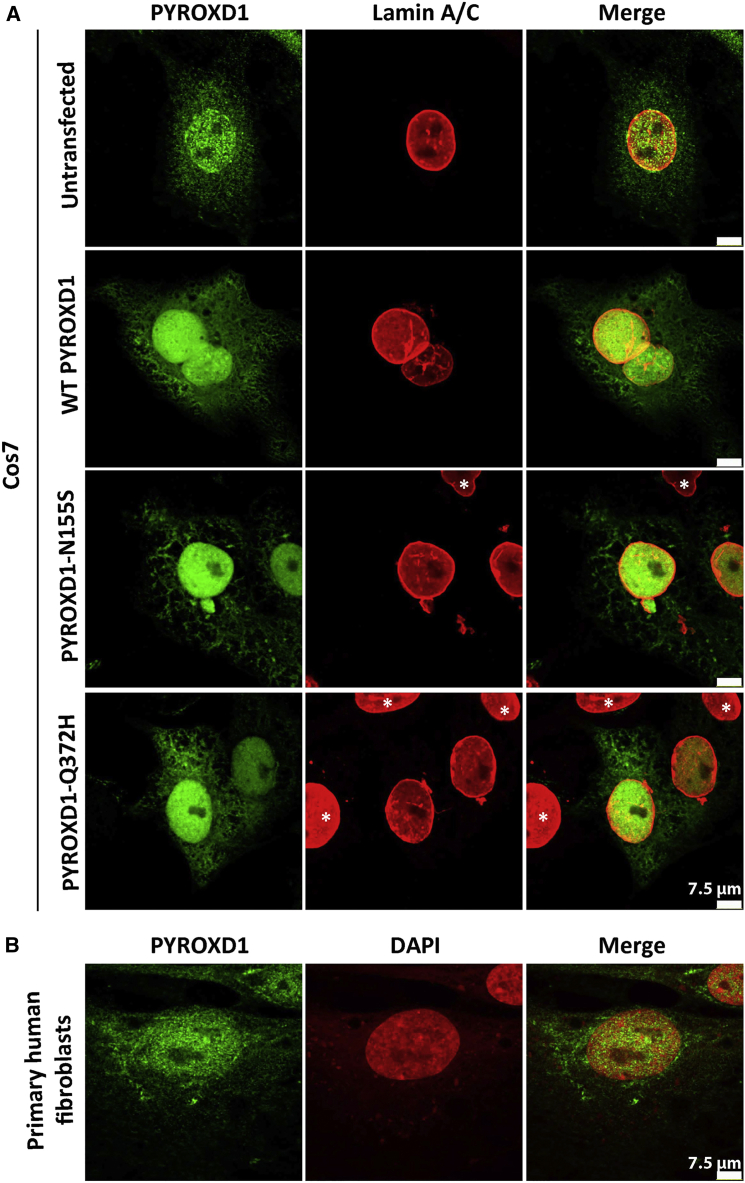
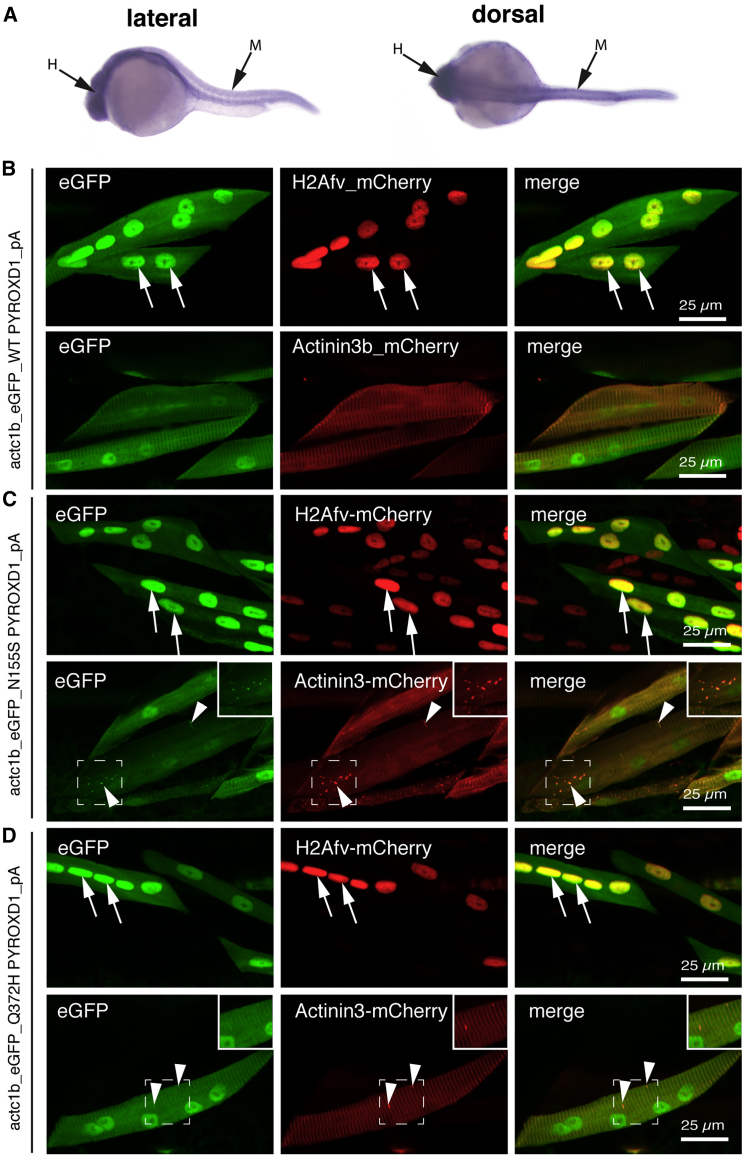
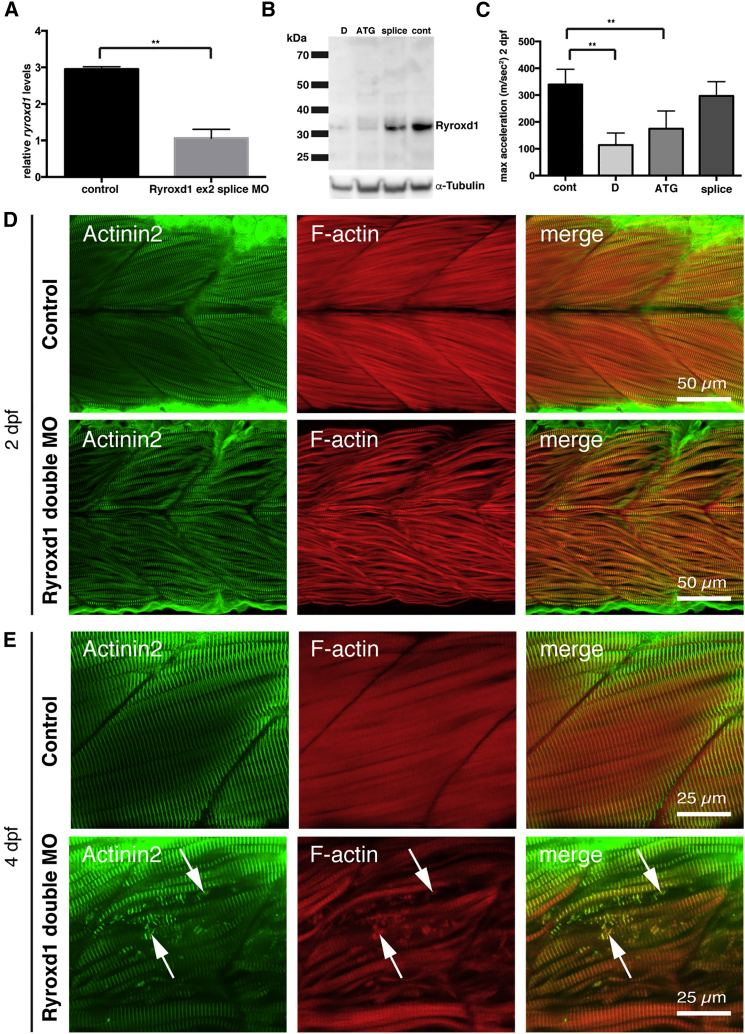
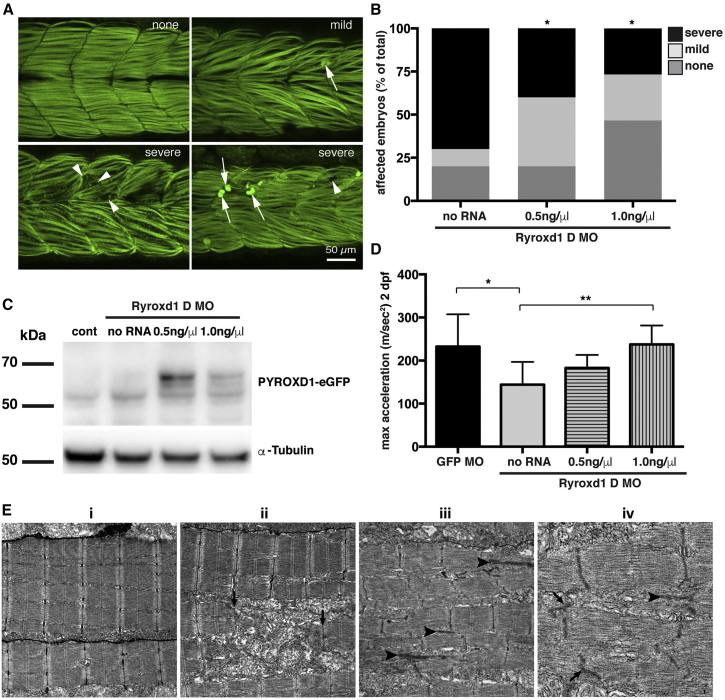
This study establishes PYROXD1 variants as a cause of early-onset myopathy and uses biospecimens and cell lines, yeast, and zebrafish models to elucidate the fundamental role of PYROXD1 in skeletal muscle. Exome sequencing identified recessive variants in PYROXD1 in nine probands from five families. Affected individuals presented in infancy or childhood with slowly progressive proximal and distal weakness, facial weakness, nasal speech, swallowing difficulties, and normal to moderately elevated creatine kinase. Distinctive histopathology showed abundant internalized nuclei, myofibrillar disorganization, desmin-positive inclusions, and thickened Z-bands. PYROXD1 is a nuclear-cytoplasmic pyridine nucleotide-disulphide reductase (PNDR). PNDRs are flavoproteins (FAD-binding) and catalyze pyridine-nucleotide-dependent (NAD/NADH) reduction of thiol residues in other proteins. Complementation experiments in yeast lacking glutathione reductase glr1 show that human PYROXD1 has reductase activity that is strongly impaired by the disease-associated missense mutations. Immunolocalization studies in human muscle and zebrafish myofibers demonstrate that PYROXD1 localizes to the nucleus and to striated sarcomeric compartments. Zebrafish with ryroxD1 knock-down recapitulate features of PYROXD1 myopathy with sarcomeric disorganization, myofibrillar aggregates, and marked swimming defect. We characterize variants in the oxidoreductase PYROXD1 as a cause of early-onset myopathy with distinctive histopathology and introduce altered redox regulation as a primary cause of congenital muscle disease.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Amburgey K., McNamara N., Bennett L.R., McCormick M.E., Acsadi G., Dowling J.J. Prevalence of congenital myopathies in a representative pediatric united states population. Ann. Neurol. 2011;70:662–665. - PubMed
-
- Chae J.H., Vasta V., Cho A., Lim B.C., Zhang Q., Eun S.H., Hahn S.H. Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders. J. Med. Genet. 2015;52:208–216. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
