

DNAJB6 Myopathies: Focused Review on an Emerging and Expanding Group of Myopathies
- PMID: 27747217
- PMCID: PMC5043021
- DOI: 10.3389/fmolb.2016.00063
DNAJB6 Myopathies: Focused Review on an Emerging and Expanding Group of Myopathies
Abstract
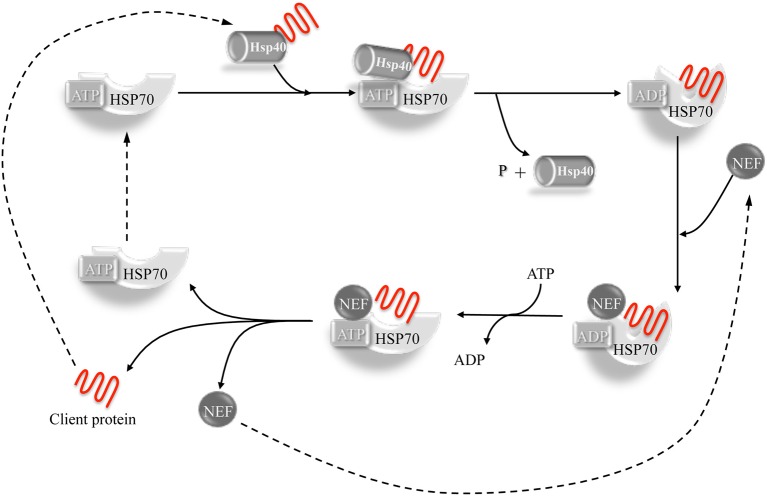
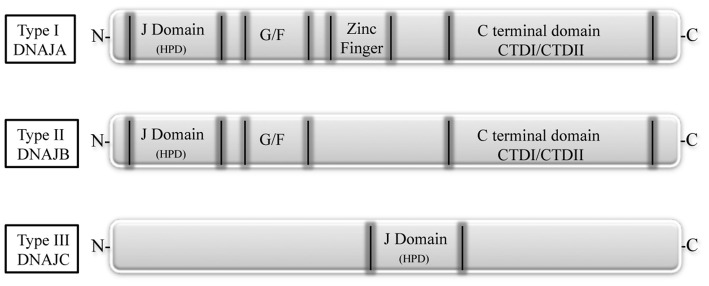
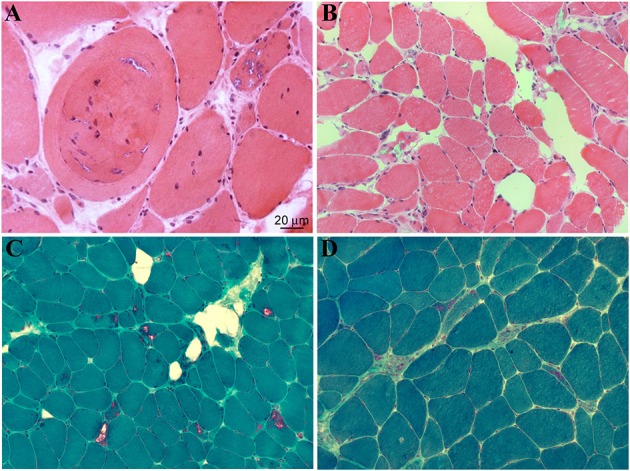
Mutations in the DNAJB6 gene have been associated with the autosomal dominant limb girdle muscular dystrophy type 1D (LGMD1D), a disorder characterized by abnormal protein aggregates and rimmed vacuoles in muscle fibers. DNAJB6 is a ubiquitously expressed Hsp40 co-chaperone characterized by a J domain that specifies Hsp70 functions in the cellular environment. DNAJB6 is also a potent inhibitor of expanded polyglutamine (polyQ) aggregation preventing aggregate toxicity in cells. In DNAJB6-mutated patients this anti-aggregation property is significantly reduced, albeit not completely lost. To elucidate the pathogenetic mechanisms underlying the DNAJB6-related myopathy, animal models have been created showing that, indeed, conditional muscular expression of a DNAJB6 mutant in the mouse causes a LGMD1D myofibrillary muscle tissue phenotype. Both mutations and phenotypes reported until recently were rather homogeneous, being exclusively missense mutations of a few amino acids of the protein G/F domain, and with a phenotype characterized by adult-onset slowly progressive muscular dystrophy predominantly affecting proximal muscles. Lately, several novel mutations and new phenotypes of DNAJB6 have been described. These mutations once more affect the G/F domain of DNAJB6 with missense changes and a splice site mutation; and the phenotypes include childhood onset and distal involvement of muscles, or childhood-onset LGMD1D with loss of ambulation in early adulthood and respiratory involvement. Thus, the spectrum of DNAJB6-related phenotypes is widening. Although our knowledge about the role of DNAJB6 in the pathogenesis of muscle diseases has made great progression, several questions remain unsolved, including why a ubiquitous protein affects only, or predominantly, skeletal muscle; why only the G/F domain is involved; and what is the possible role of the DNAJB6a isoform. Clarification of these issues will provide clues to implement possible therapeutic strategies for DNAJB6-related myopathies.
Keywords: DNAJB6; LGMD1D; autophagy; chaperone; distal myopathy; protein aggregation; vacuolar myopathy.
Figures

References
-
- Anfinsen C. B. (1973). Principles that govern the folding of protein chains. Science 181, 223–230. - PubMed
-
- Couthouis J., Raphael A. R., Siskind C., Findlay A. R., Buenrostro J. D., Greenleaf W. J., et al. . (2014). Exome sequencing identifies a DNAJB6 mutation in a family with dominantly-inherited limb-girdle muscular dystrophy. Neuromuscul. Disord. 24, 431–435. 10.1016/j.nmd.2014.01.014 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources