

ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing
- PMID: 27749837
- PMCID: PMC5509561
- DOI: 10.1038/nmeth.4031
ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing
Abstract
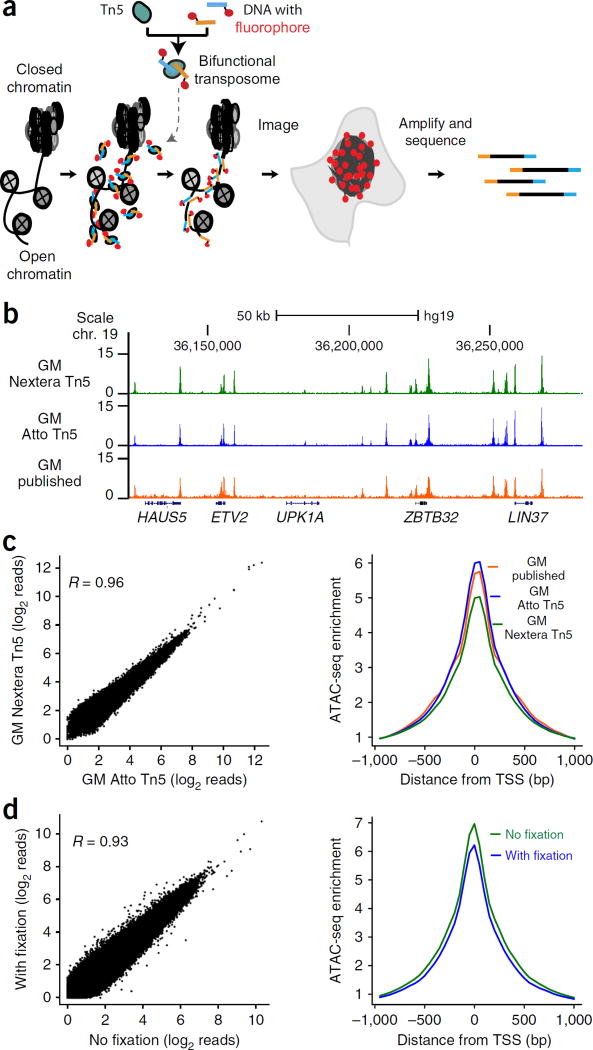
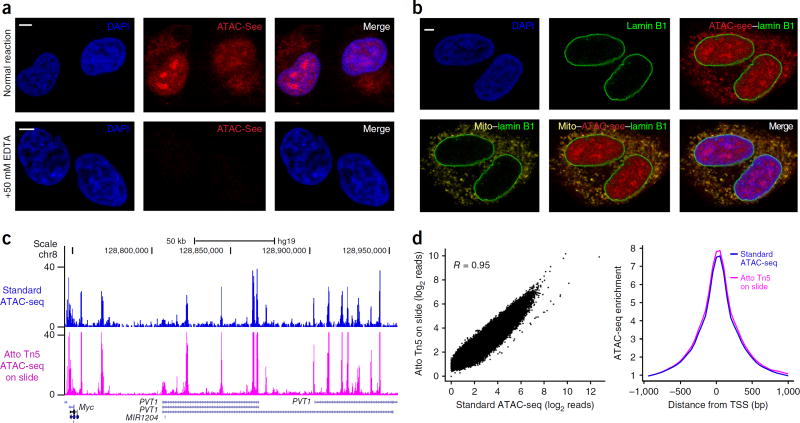
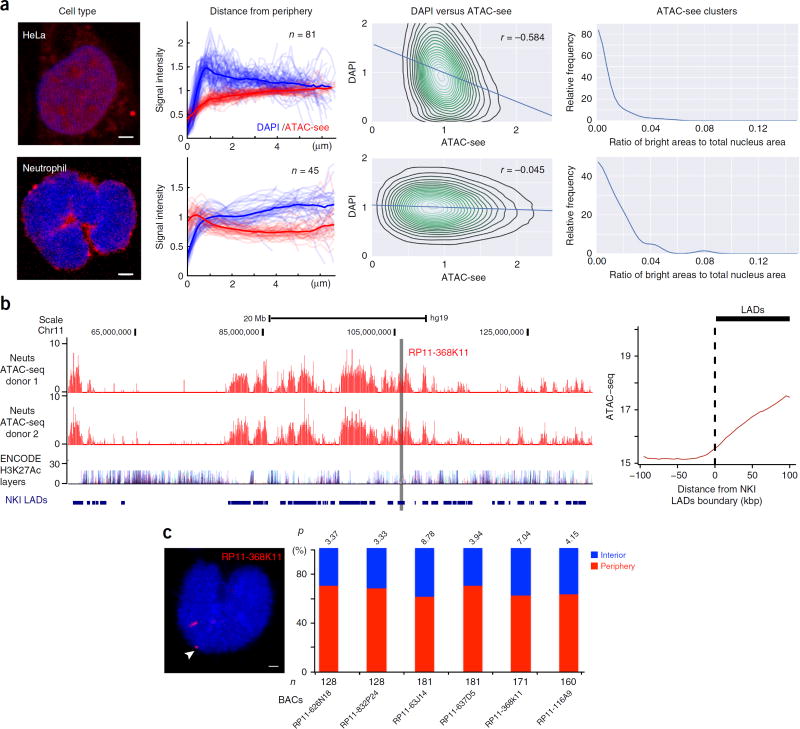
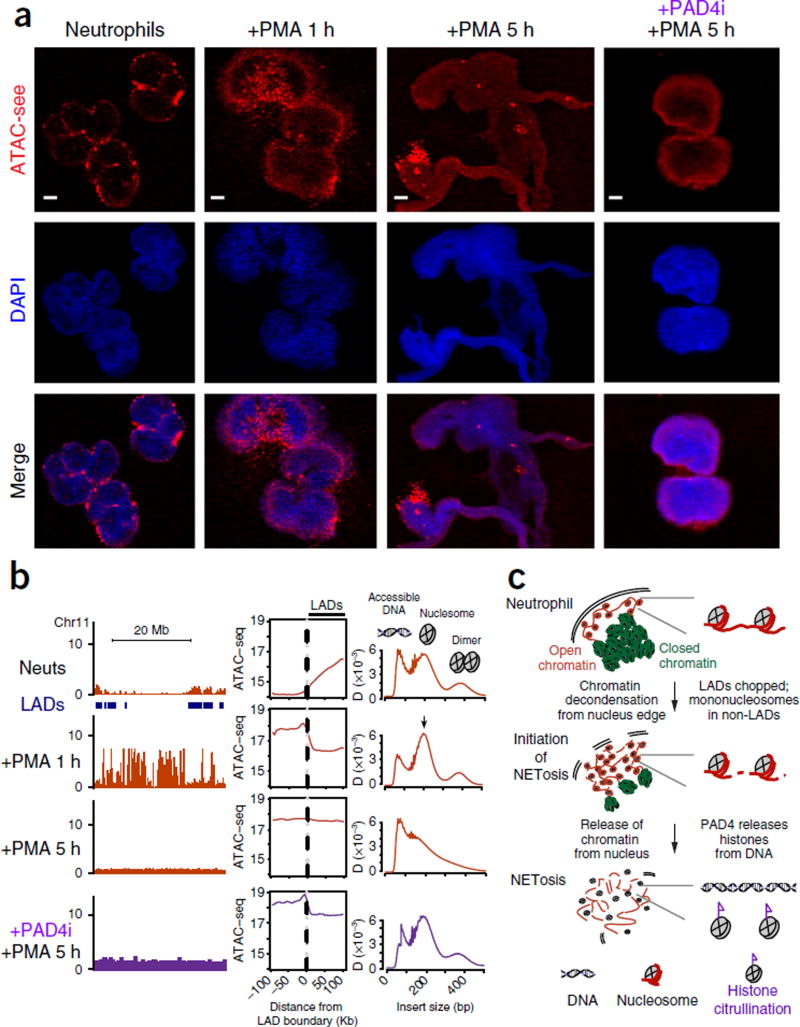
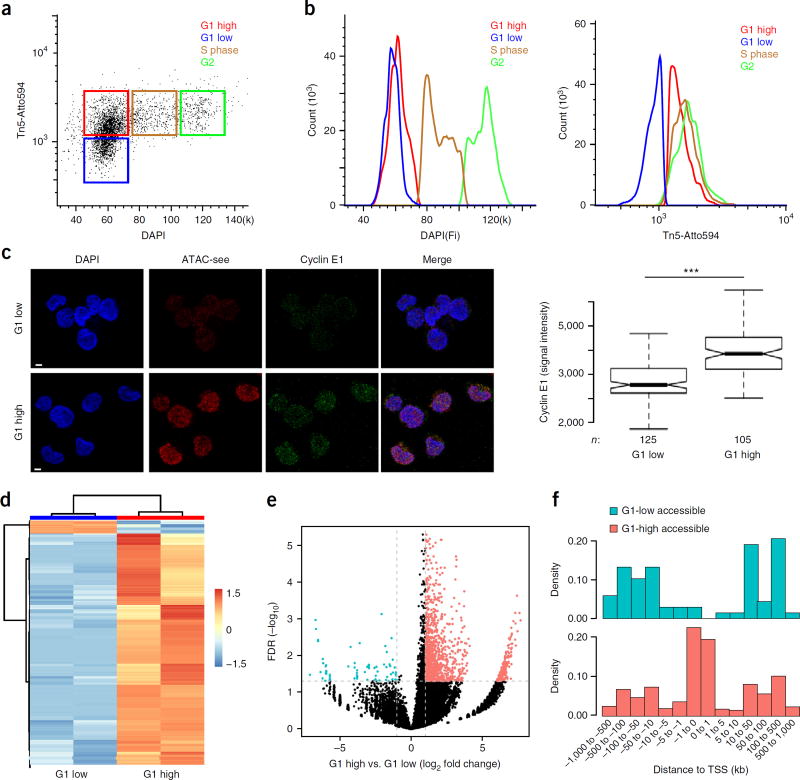
Spatial organization of the genome plays a central role in gene expression, DNA replication, and repair. But current epigenomic approaches largely map DNA regulatory elements outside of the native context of the nucleus. Here we report assay of transposase-accessible chromatin with visualization (ATAC-see), a transposase-mediated imaging technology that employs direct imaging of the accessible genome in situ, cell sorting, and deep sequencing to reveal the identity of the imaged elements. ATAC-see revealed the cell-type-specific spatial organization of the accessible genome and the coordinated process of neutrophil chromatin extrusion, termed NETosis. Integration of ATAC-see with flow cytometry enables automated quantitation and prospective cell isolation as a function of chromatin accessibility, and it reveals a cell-cycle dependence of chromatin accessibility that is especially dynamic in G1 phase. The integration of imaging and epigenomics provides a general and scalable approach for deciphering the spatiotemporal architecture of gene control.
Conflict of interest statement
The authors declare competing financial interests: details are available in the
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
