

Prospective functional classification of all possible missense variants in PPARG
- PMID: 27749844
- PMCID: PMC5131844
- DOI: 10.1038/ng.3700
Prospective functional classification of all possible missense variants in PPARG
Abstract
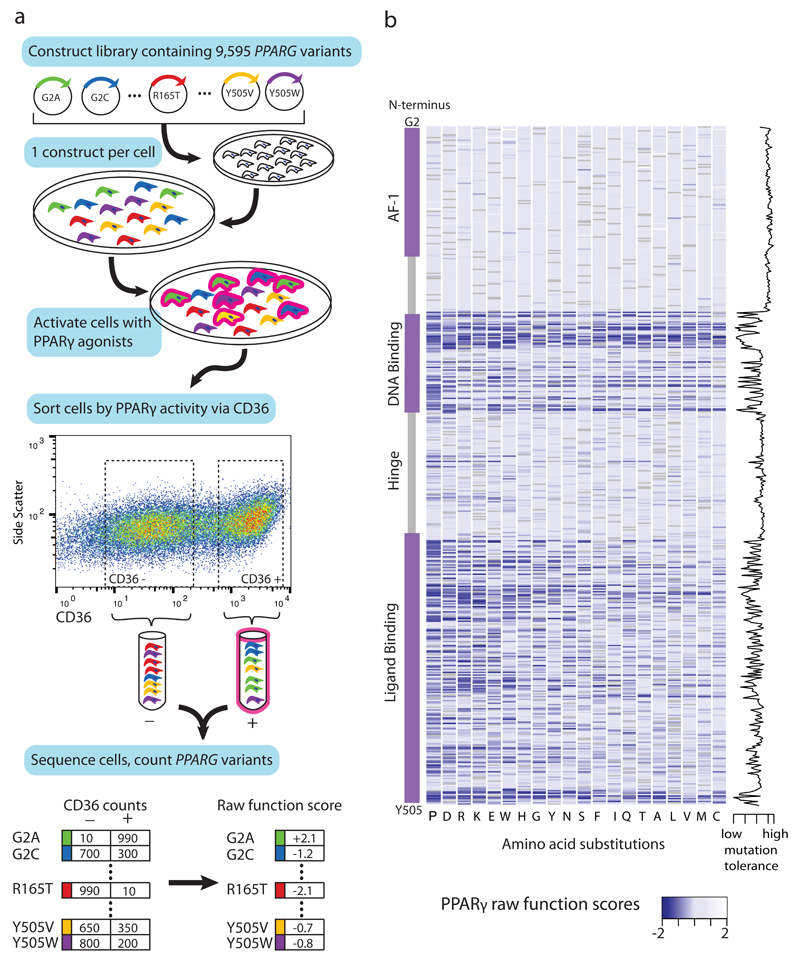
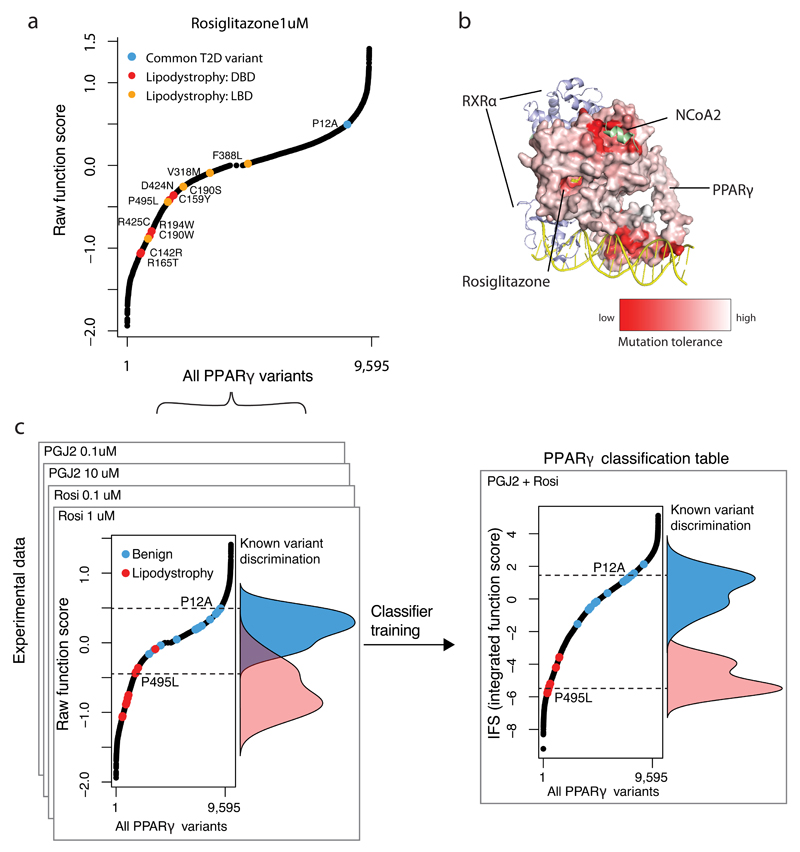
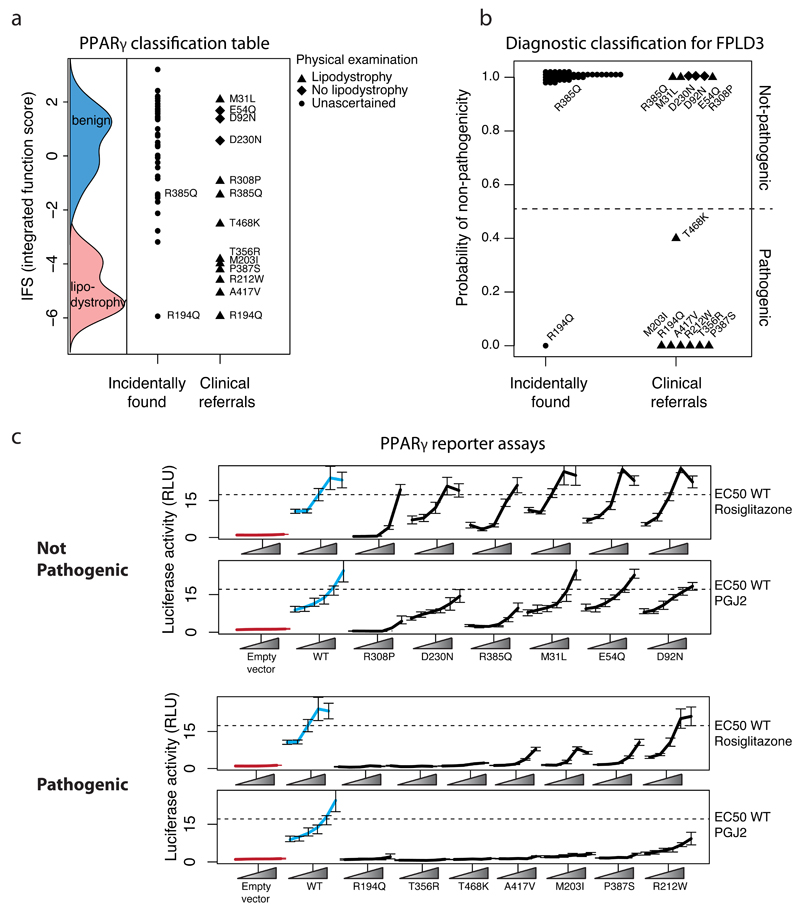
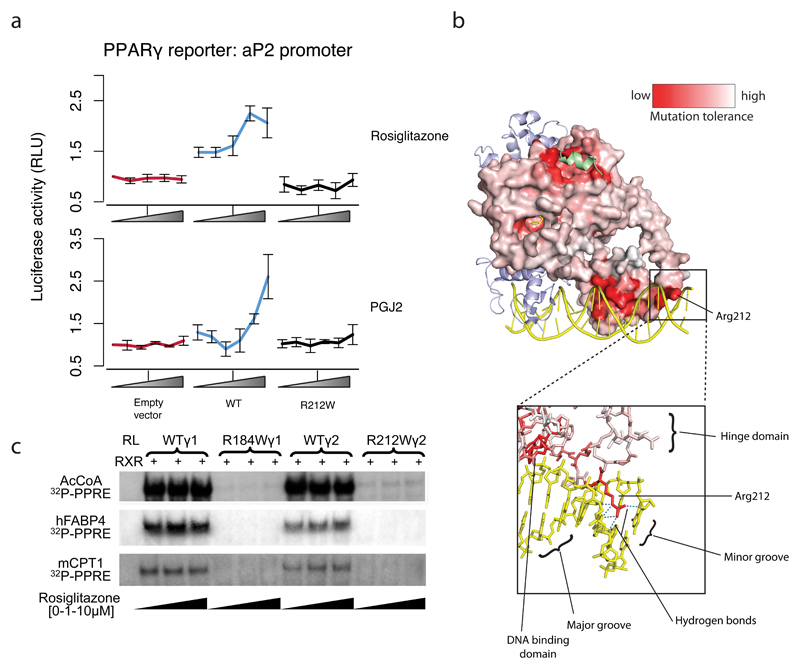
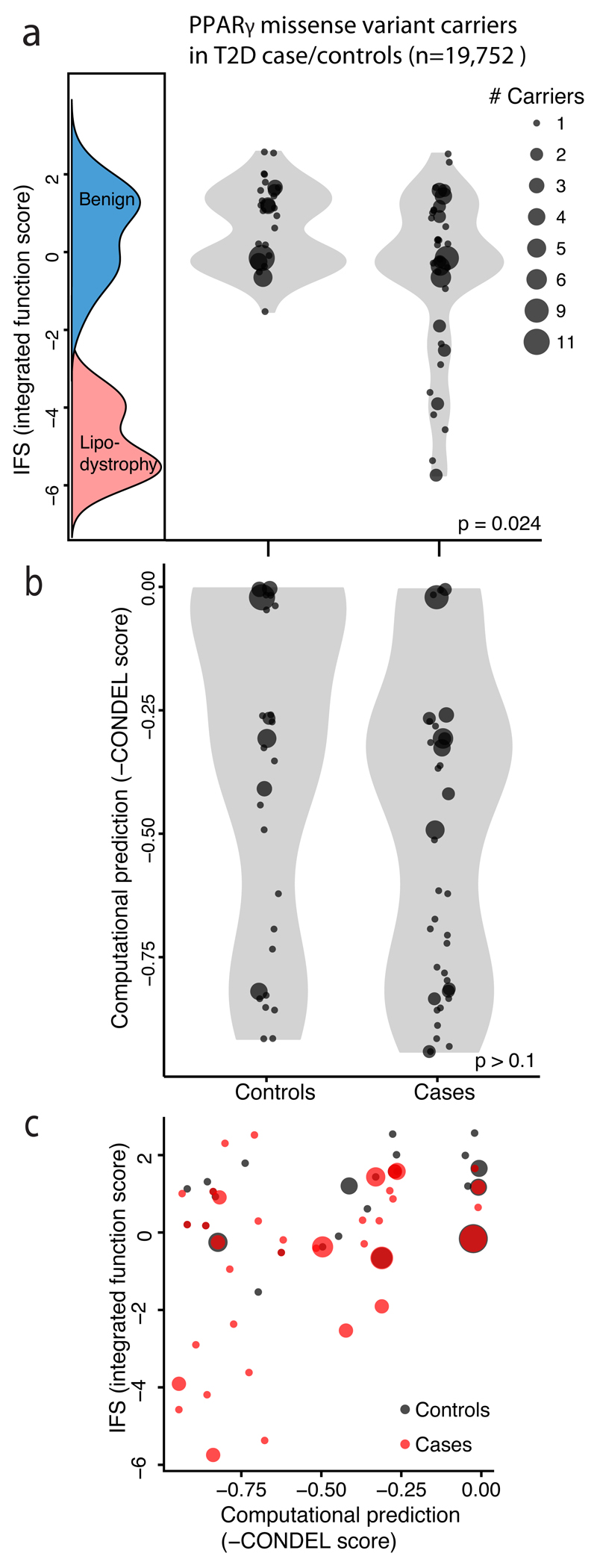
Clinical exome sequencing routinely identifies missense variants in disease-related genes, but functional characterization is rarely undertaken, leading to diagnostic uncertainty. For example, mutations in PPARG cause Mendelian lipodystrophy and increase risk of type 2 diabetes (T2D). Although approximately 1 in 500 people harbor missense variants in PPARG, most are of unknown consequence. To prospectively characterize PPARγ variants, we used highly parallel oligonucleotide synthesis to construct a library encoding all 9,595 possible single-amino acid substitutions. We developed a pooled functional assay in human macrophages, experimentally evaluated all protein variants, and used the experimental data to train a variant classifier by supervised machine learning. When applied to 55 new missense variants identified in population-based and clinical sequencing, the classifier annotated 6 variants as pathogenic; these were subsequently validated by single-variant assays. Saturation mutagenesis and prospective experimental characterization can support immediate diagnostic interpretation of newly discovered missense variants in disease-related genes.
Conflict of interest statement
No competing financial interests
Figures
References
-
- Majewski J, Schwartzentruber J, Lalonde E, Montpetit A, Jabado N. What can exome sequencing do for you? J Med Genet. 2011;48:580–9. - PubMed
-
- Barroso I, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–3. - PubMed
-
- Jeninga E, Gurnell M. Functional implications of genetic variation in human PPAR [gamma] Trends in Endocrinology & …. 2009 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
