

PlexinD1 Is a Novel Transcriptional Target and Effector of Notch Signaling in Cancer Cells
- PMID: 27749937
- PMCID: PMC5066946
- DOI: 10.1371/journal.pone.0164660
PlexinD1 Is a Novel Transcriptional Target and Effector of Notch Signaling in Cancer Cells
Erratum in
-
Correction: PlexinD1 Is a Novel Transcriptional Target and Effector of Notch Signaling in Cancer Cells.PLoS One. 2016 Dec 9;11(12):e0168429. doi: 10.1371/journal.pone.0168429. eCollection 2016. PLoS One. 2016. PMID: 27936215 Free PMC article.
Abstract
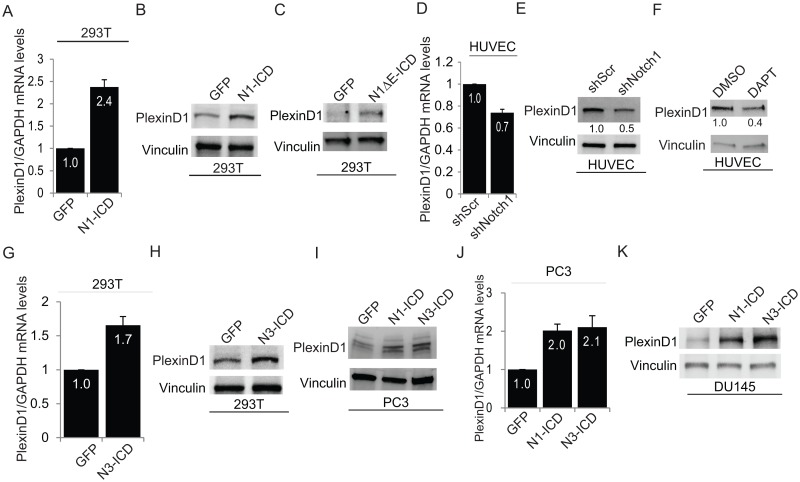
The secreted semaphorin Sema3E controls cell migration and invasiveness in cancer cells. Sema3E-receptor, PlexinD1, is frequently upregulated in melanoma, breast, colon, ovarian and prostate cancers; however, the mechanisms underlying PlexinD1 upregulation and the downstream events elicited in tumor cells are still unclear. Here we show that the canonical RBPjk-dependent Notch signaling cascade controls PlexinD1 expression in primary endothelial and cancer cells. Transcriptional activation was studied by quantitative PCR and promoter activity reporter assays. We found that Notch ligands and constitutively activated intracellular forms of Notch receptors upregulated PlexinD1 expression; conversely RNAi-based knock-down, or pharmacological inhibition of Notch signaling by gamma-secretase inhibitors, downregulated PlexinD1 levels. Notably, both Notch1 and Notch3 expression positively correlates with PlexinD1 levels in prostate cancer, as well as in other tumor types. In prostate cancer cells, Sema3E-PlexinD1 axis was previously reported to regulate migration; however, implicated mechanisms were not elucidated. Here we show that in these cells PlexinD1 activity induces the expression of the transcription factor Slug, downregulates E-cadherin levels and enhances cell migration. Moreover, our mechanistic data identify PlexinD1 as a pivotal mediator of this signaling axis downstream of Notch in prostate cancer cells. In fact, on one hand, PlexinD1 is required to mediate cell migration and E-cadherin regulation elicited by Notch. On the other hand, PlexinD1 upregulation is sufficient to induce prostate cancer cell migration and metastatic potential in mice, leading to functional rescue in the absence of Notch. In sum, our work identifies PlexinD1 as a novel transcriptional target induced by Notch signaling, and reveals its role promoting prostate cancer cell migration and downregulating E-cadherin levels in Slug-dependent manner. Collectively, these findings suggest that Notch-PlexinD1 signaling axis may be targeted to impair prostate cancer cell invasiveness and metastasis.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





References
-
- Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, et al. (1999) Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99: 71–80. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
