

LncRNA SRA promotes hepatic steatosis through repressing the expression of adipose triglyceride lipase (ATGL)
- PMID: 27759039
- PMCID: PMC5069493
- DOI: 10.1038/srep35531
LncRNA SRA promotes hepatic steatosis through repressing the expression of adipose triglyceride lipase (ATGL)
Abstract
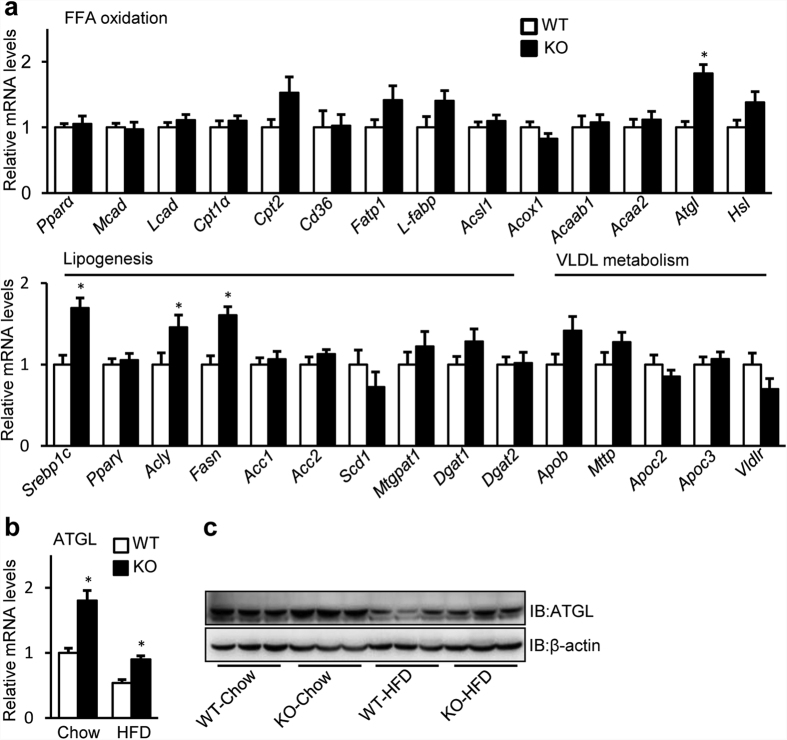
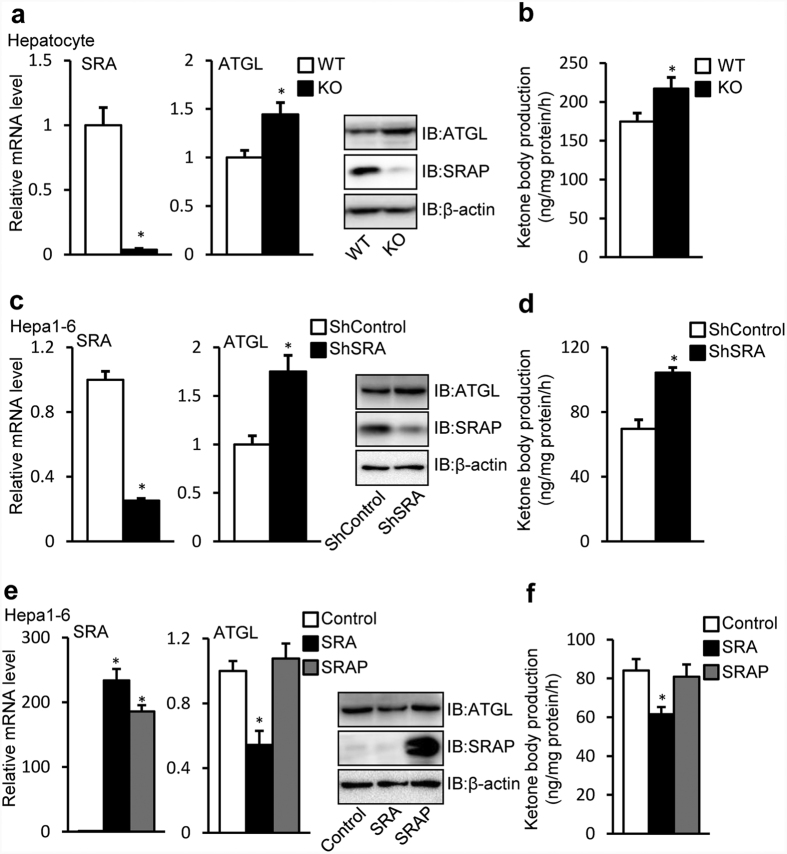
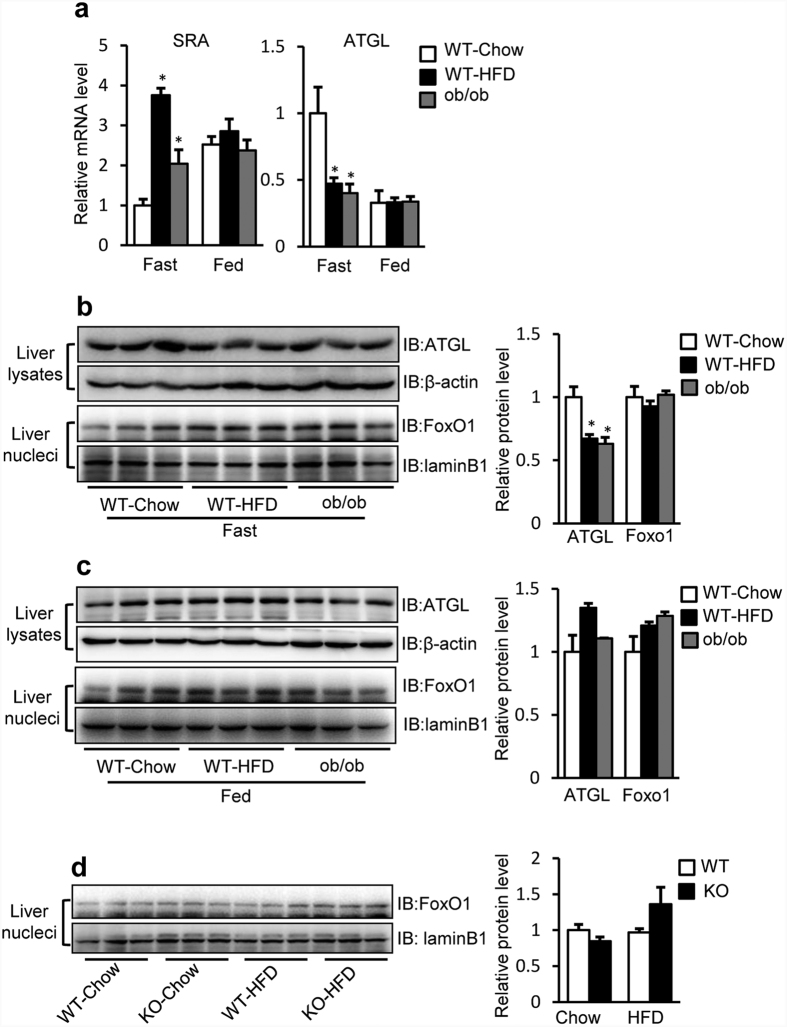
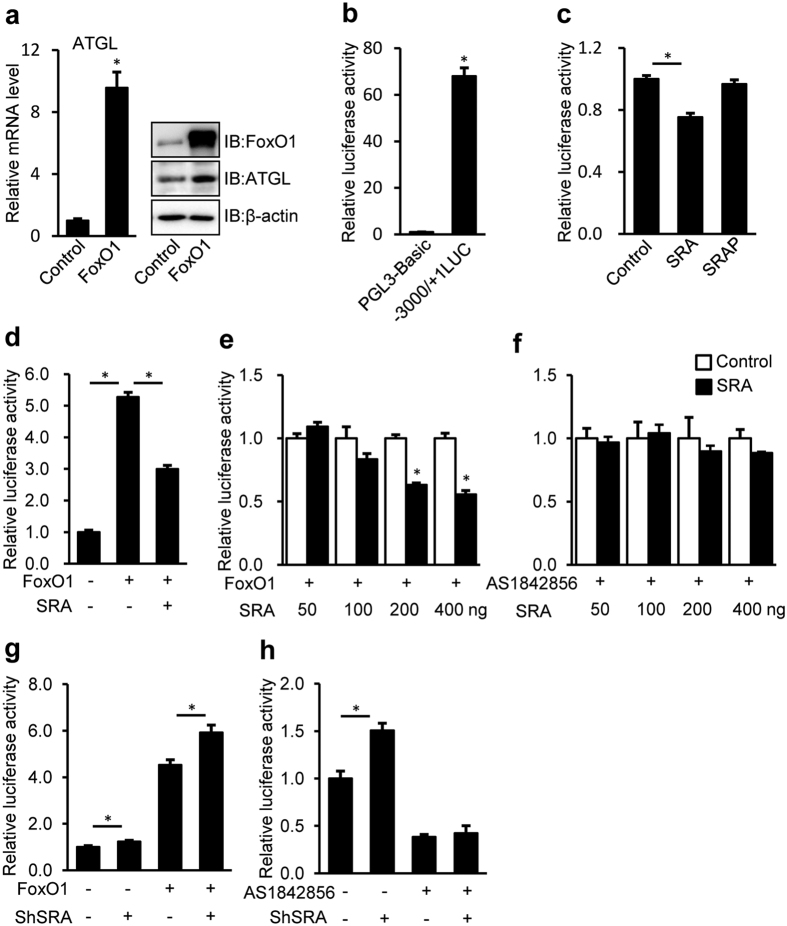
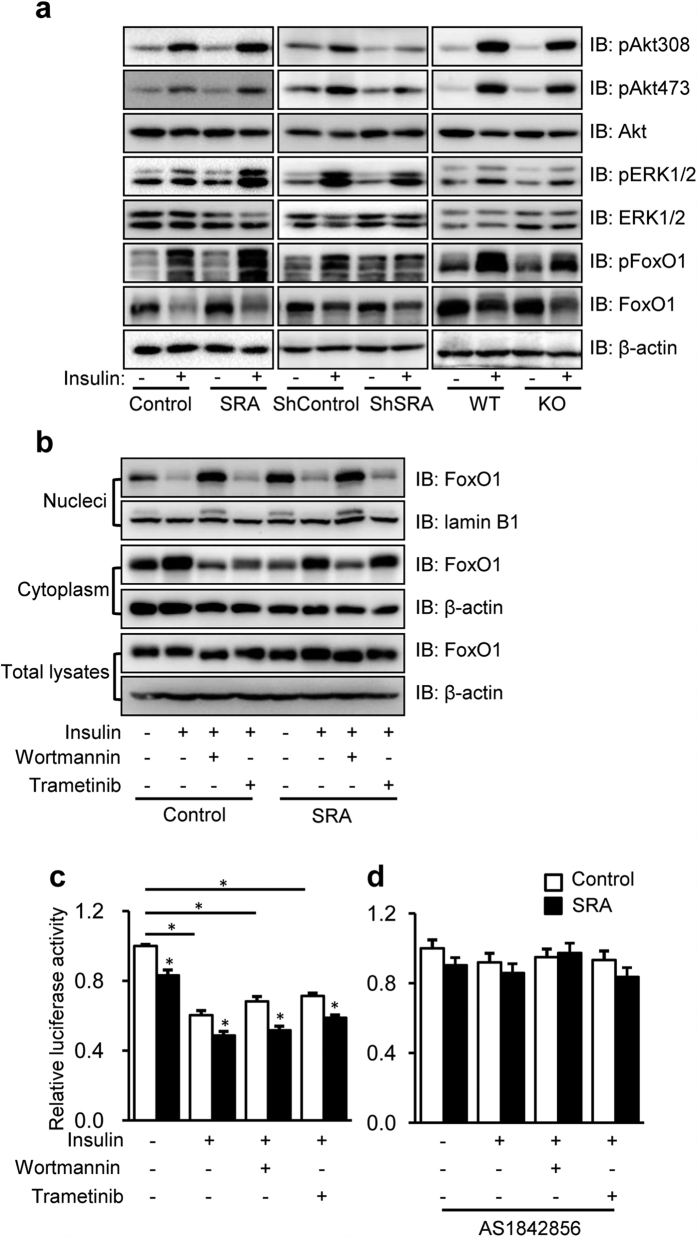
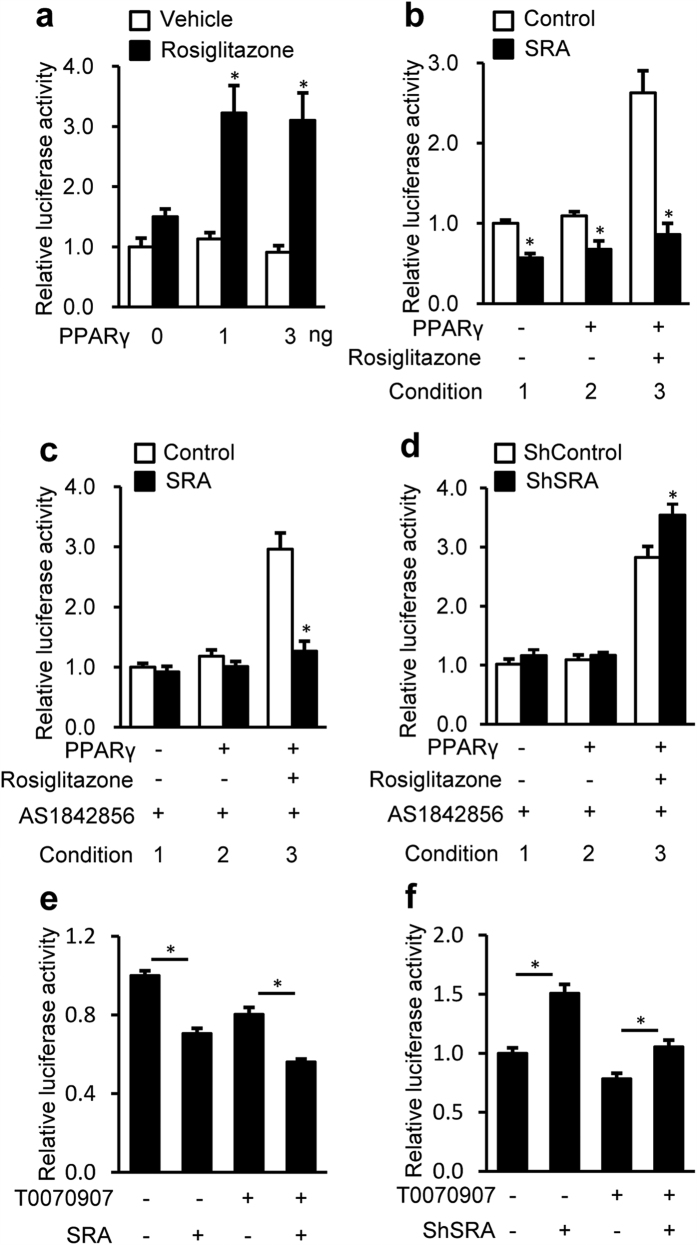
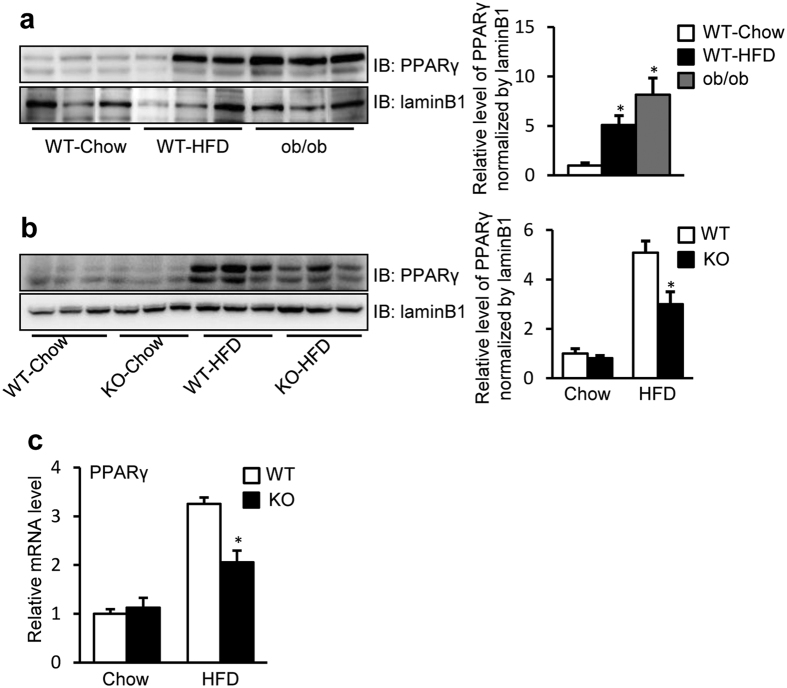
Nonalcoholic fatty liver disease (NAFLD), the most common form of chronic liver disease, manifests as an over-accumulation of hepatic fat. We have recently shown that mice with genetic knockout of a long non-coding RNA (lncRNA) steroid receptor RNA activator (SRA) (SRAKO) are resistant to high fat diet-induced obesity with a phenotype that includes improved glucose tolerance and attenuated hepatic steatosis. The underlying mechanism was investigated in the present study. We found that hepatic levels of SRA and adipose triglyceride lipase (ATGL), a major hepatic triacylglycerol (TAG) hydrolase, were inversely regulated by fasting in mice, and the expression of liver ATGL was induced by SRAKO under normal and high fat diet (HFD) feeding. Loss of SRA in primary hepatocytes or a hepatocyte cell line upregulates, but forced expression of SRA inhibits ATGL expression and free fatty acids (FFA) β-oxidation. SRA inhibits ATGL promoter activity, primarily by inhibiting the otherwise-inductive effects of the transcription factor, forkhead box protein O1 (FoxO1). Our data reveal a novel function of SRA in promoting hepatic steatosis through repression of ATGL expression.
Figures
References
-
- Marchesini G. et al.. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 50, 1844–1850 (2001). - PubMed
-
- Youssef W. & Mccullough A. J. Diabetes mellitus, obesity, and hepatic steatosis. Seminars in Gastrointestinal Disease 13, 17–30 (2002). - PubMed
-
- Rinella M. E. Nonalcoholic fatty liver disease: a systematic review. Jama the Journal of the American Medical Association 313, 2263–2273 (2015). - PubMed
-
- Ruhl C. E. & Everhart J. E. Fatty liver indices in the multiethnic United States National Health and Nutrition Examination Survey. Alimentary Pharmacology & Therapeutics 41, 65–76 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
