

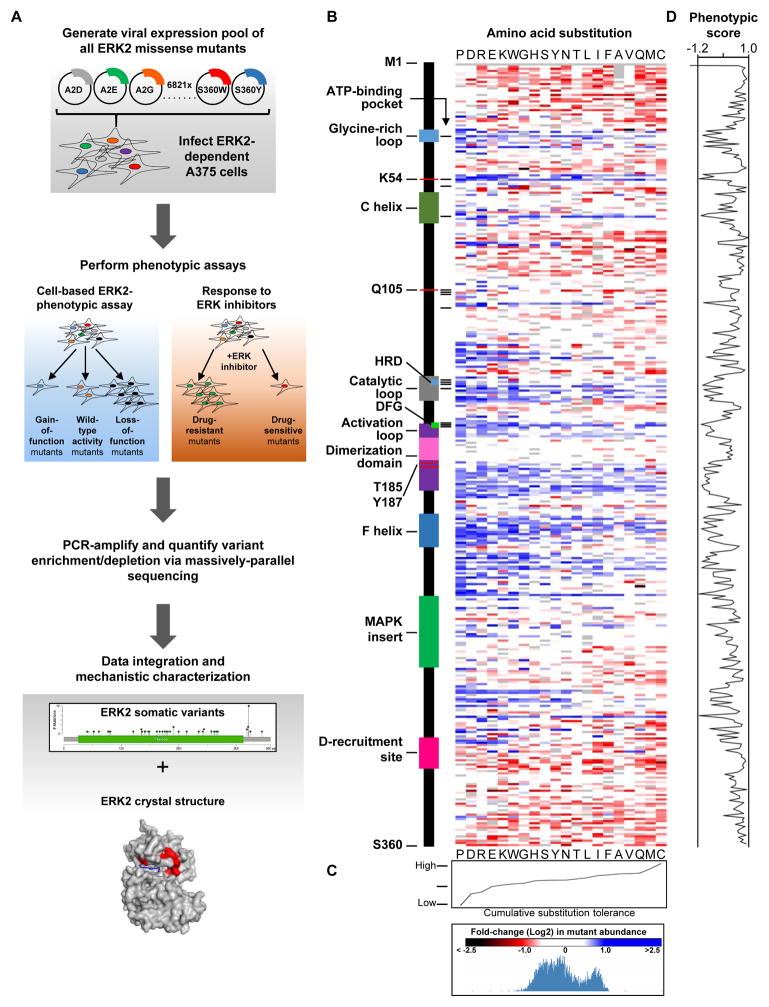
Phenotypic Characterization of a Comprehensive Set of MAPK1/ERK2 Missense Mutants
- PMID: 27760319
- PMCID: PMC5120861
- DOI: 10.1016/j.celrep.2016.09.061
Phenotypic Characterization of a Comprehensive Set of MAPK1/ERK2 Missense Mutants
Abstract
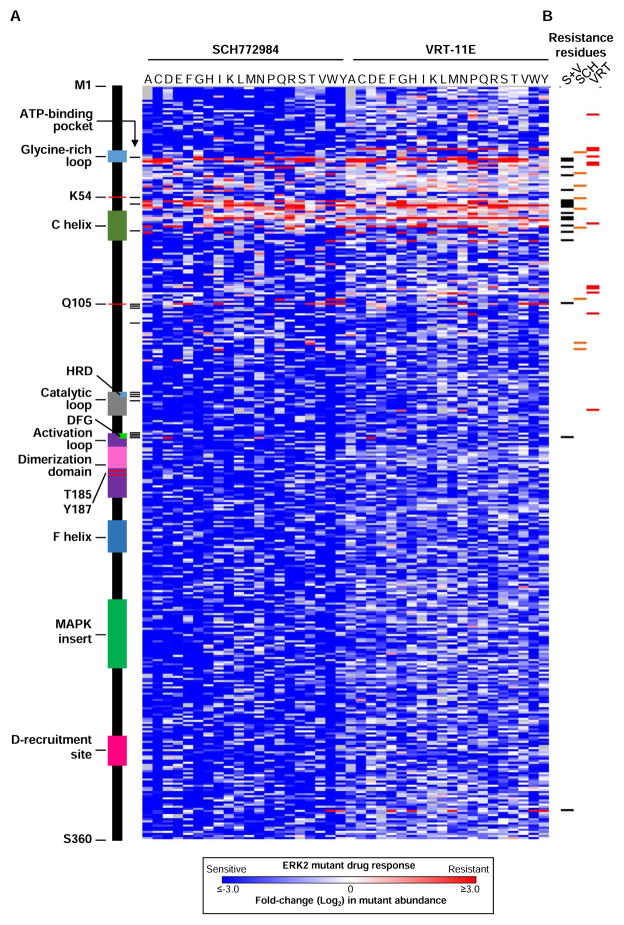
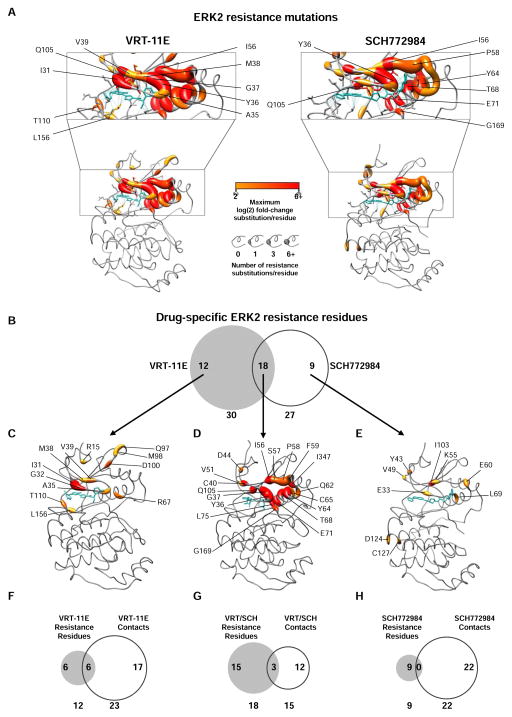
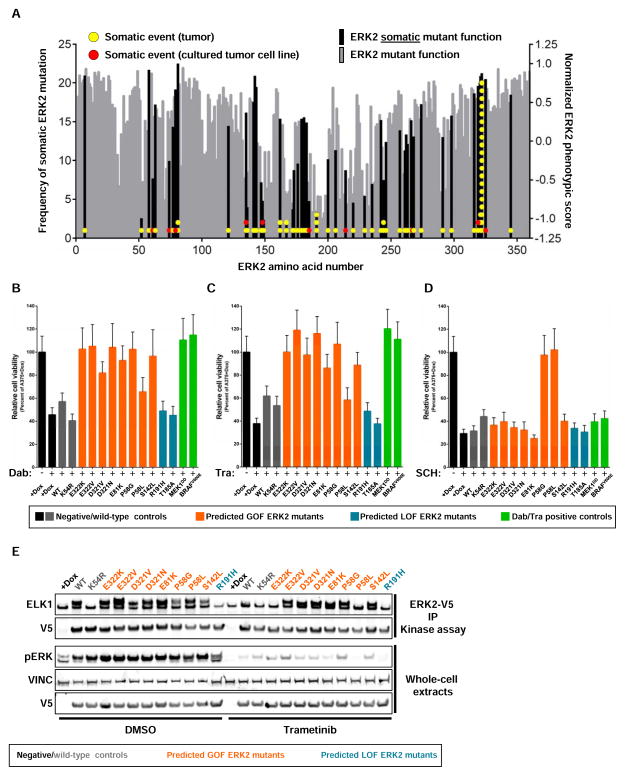
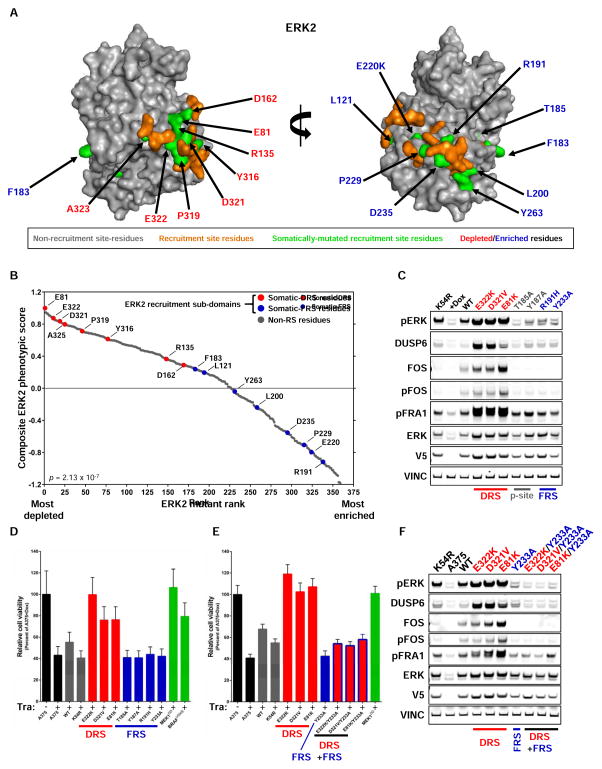
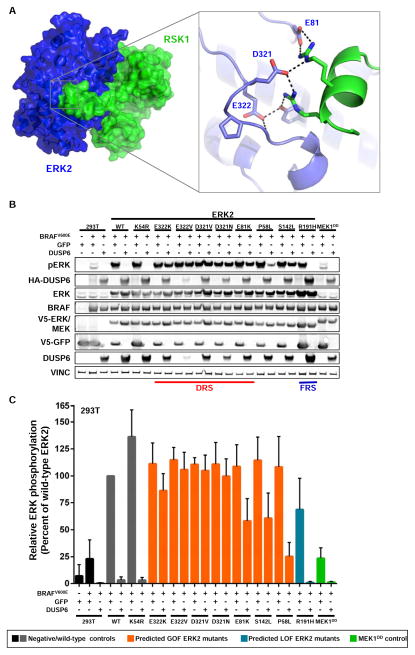
Tumor-specific genomic information has the potential to guide therapeutic strategies and revolutionize patient treatment. Currently, this approach is limited by an abundance of disease-associated mutants whose biological functions and impacts on therapeutic response are uncharacterized. To begin to address this limitation, we functionally characterized nearly all (99.84%) missense mutants of MAPK1/ERK2, an essential effector of oncogenic RAS and RAF. Using this approach, we discovered rare gain- and loss-of-function ERK2 mutants found in human tumors, revealing that, in the context of this assay, mutational frequency alone cannot identify all functionally impactful mutants. Gain-of-function ERK2 mutants induced variable responses to RAF-, MEK-, and ERK-directed therapies, providing a reference for future treatment decisions. Tumor-associated mutations spatially clustered in two ERK2 effector-recruitment domains yet produced mutants with opposite phenotypes. This approach articulates an allele-characterization framework that can be scaled to meet the goals of genome-guided oncology.
Keywords: ERK; MAPK; cancer; functional biology; precision medicine; precision oncology; rare mutants.
Copyright © 2016 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Aronov AM, Tang Q, Martinez-Botella G, Bemis GW, Cao J, Chen G, Ewing NP, Ford PJ, Germann UA, Green J, et al. Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control. J Med Chem. 2009;52:6362–6368. - PubMed
-
- Bott CM, Thorneycroft SG, Marshall CJ. The sevenmaker gain-of-function mutation in p42 MAP kinase leads to enhanced signalling and reduced sensitivity to dual specificity phosphatase action. FEBS Lett. 1994;352:201–205. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
