

Deletion of protein kinase C-ε attenuates mitochondrial dysfunction and ameliorates ischemic renal injury
- PMID: 27760765
- PMCID: PMC5283890
- DOI: 10.1152/ajprenal.00115.2016
Deletion of protein kinase C-ε attenuates mitochondrial dysfunction and ameliorates ischemic renal injury
Abstract
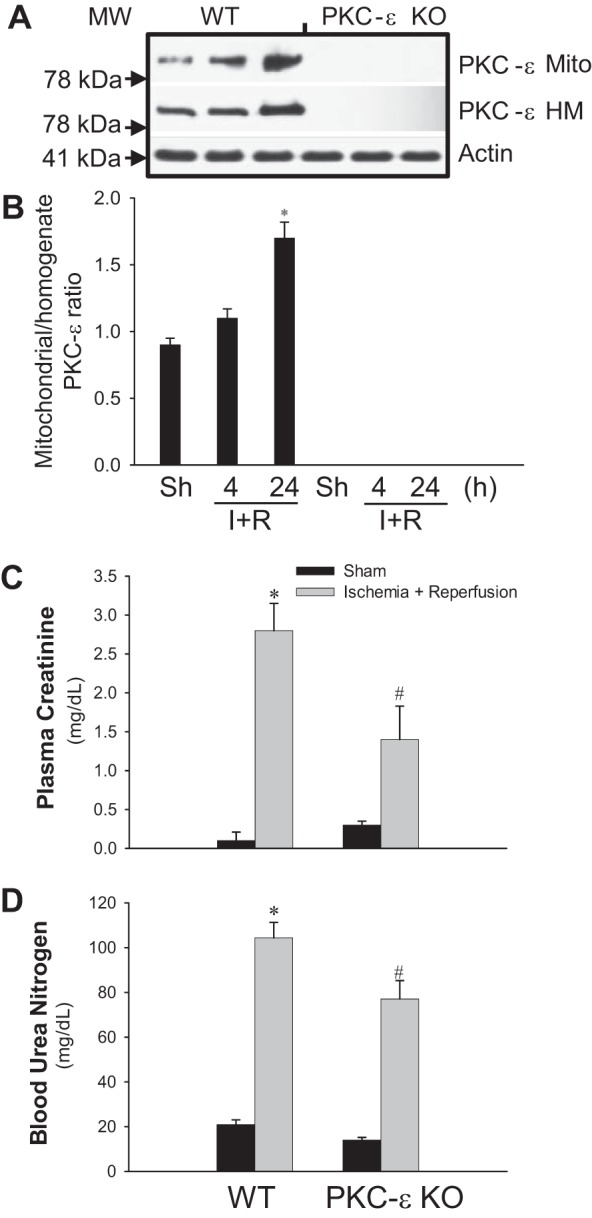
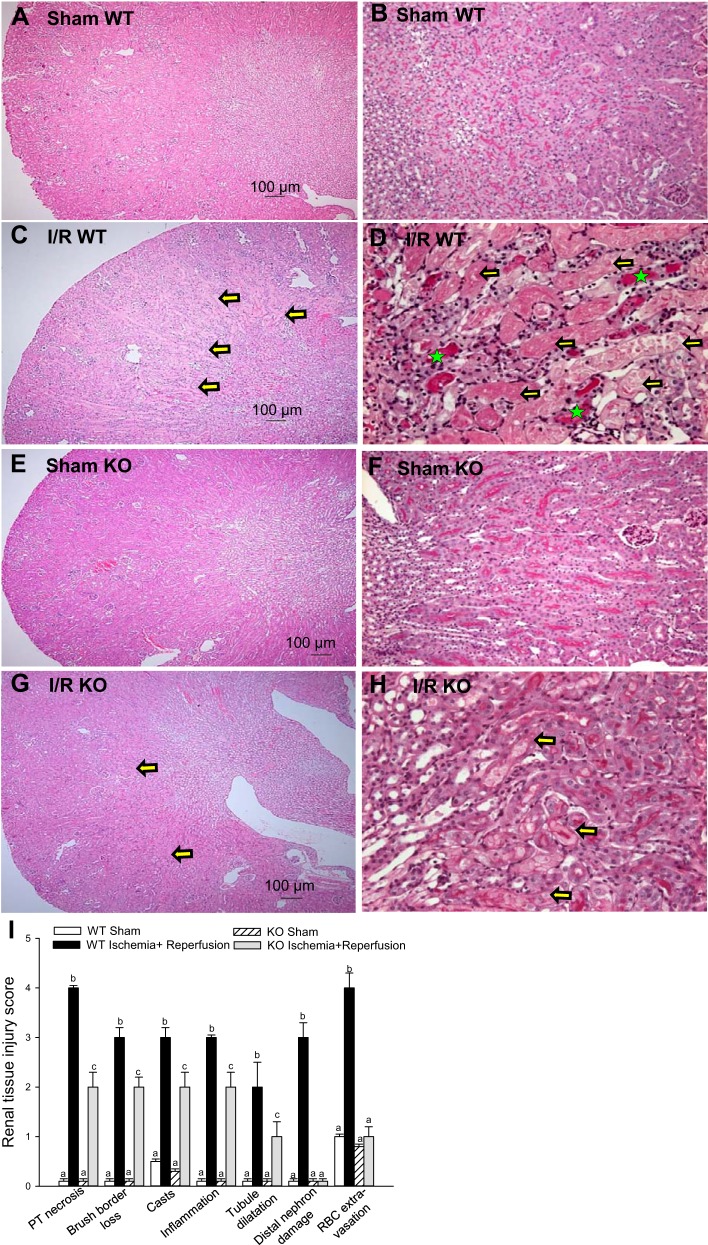
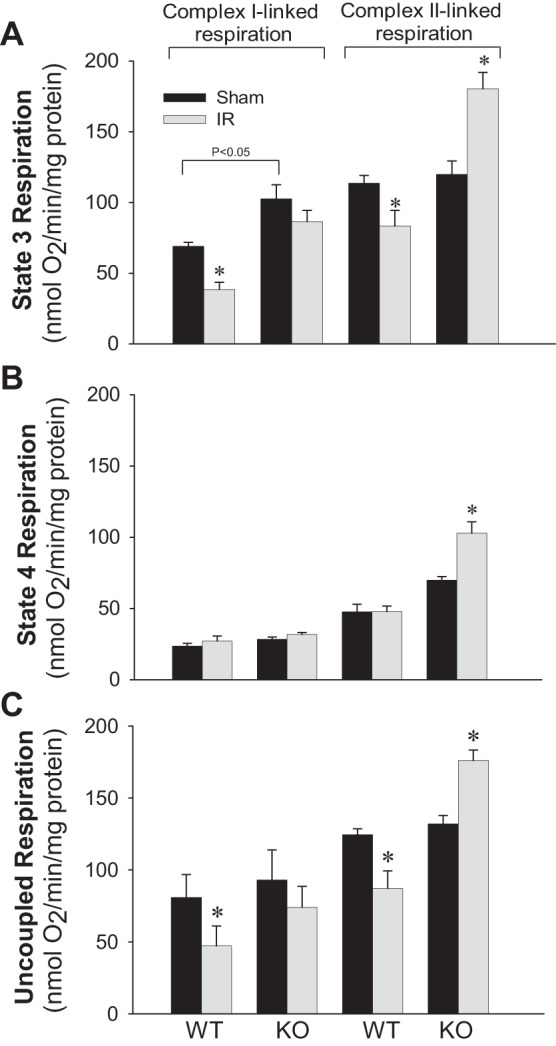
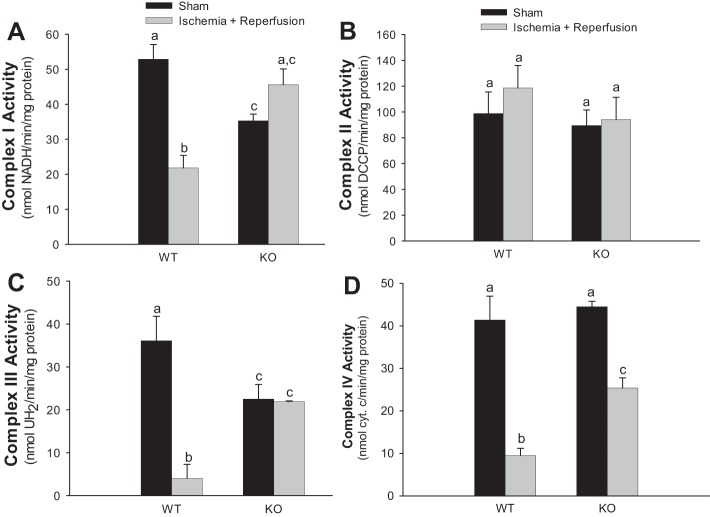
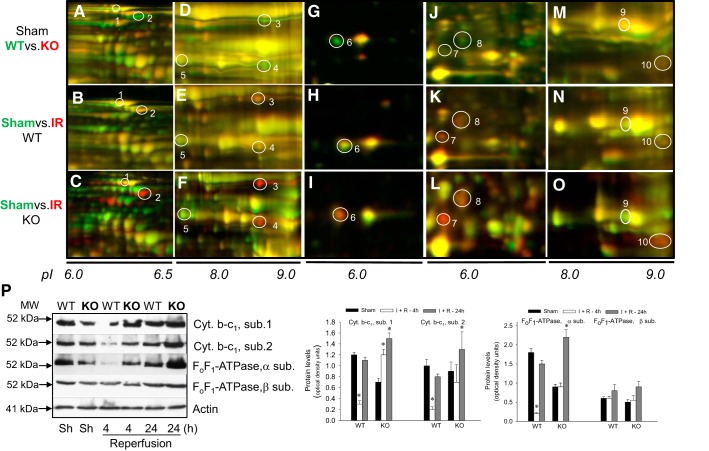
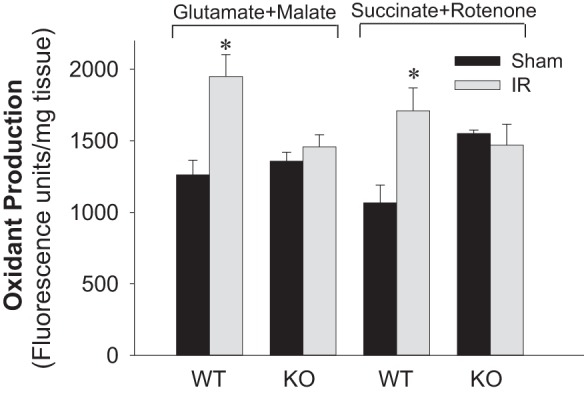
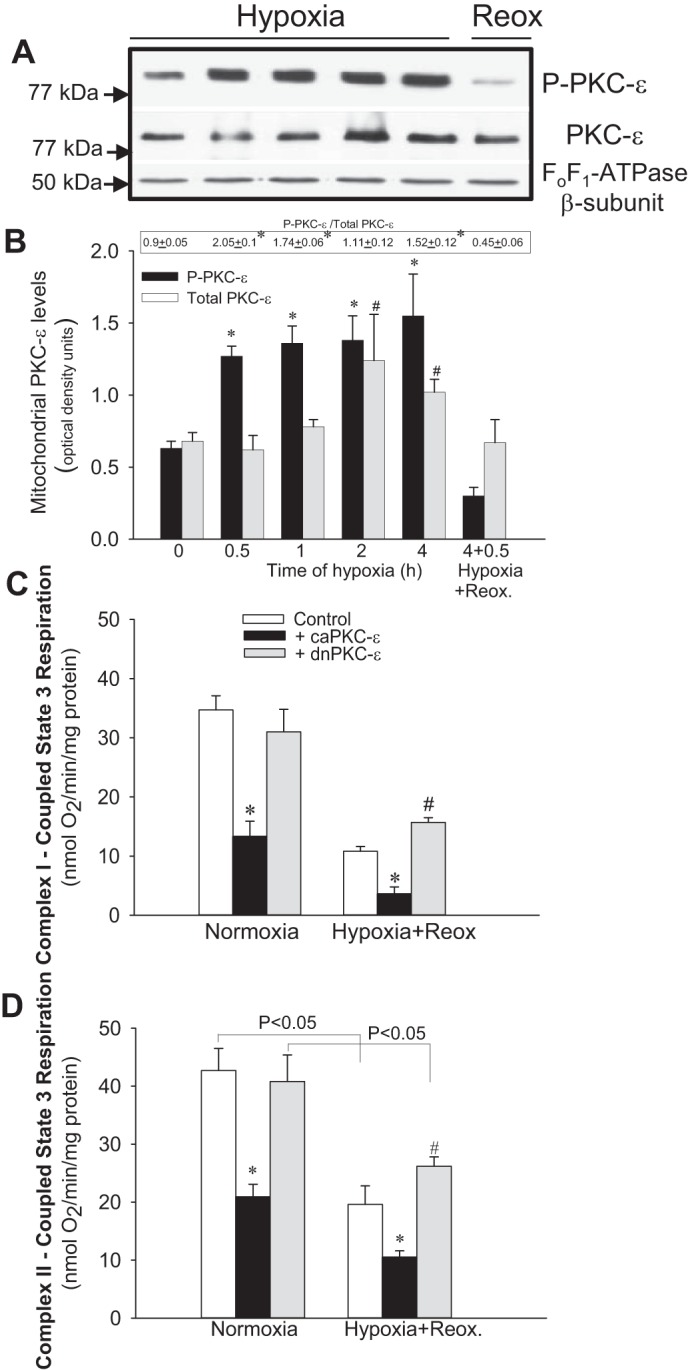
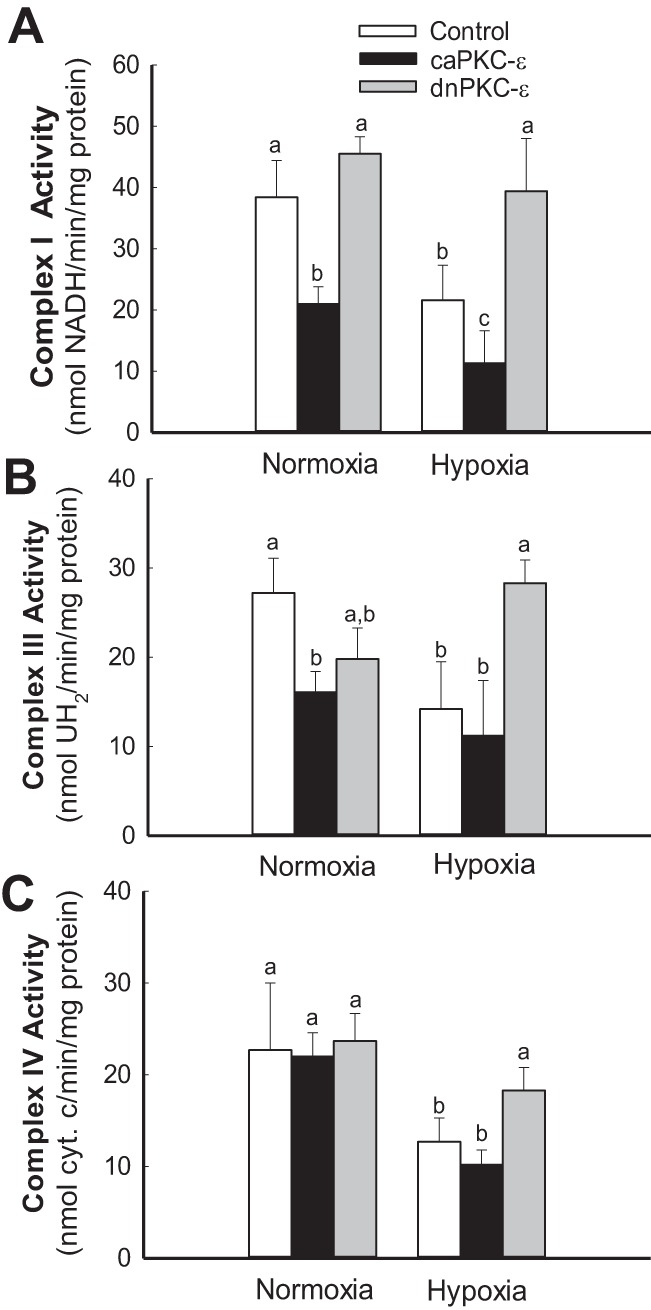
Previously, we documented that activation of protein kinase C-ε (PKC-ε) mediates mitochondrial dysfunction in cultured renal proximal tubule cells (RPTC). This study tested whether deletion of PKC-ε decreases dysfunction of renal cortical mitochondria and improves kidney function after renal ischemia. PKC-ε levels in mitochondria of ischemic kidneys increased 24 h after ischemia. Complex I- and complex II-coupled state 3 respirations were reduced 44 and 27%, respectively, in wild-type (WT) but unchanged and increased in PKC-ε-deficient (KO) mice after ischemia. Respiratory control ratio coupled to glutamate/malate oxidation decreased 50% in WT but not in KO mice. Activities of complexes I, III, and IV were decreased 59, 89, and 61%, respectively, in WT but not in KO ischemic kidneys. Proteomics revealed increases in levels of ATP synthase (α-subunit), complexes I and III, cytochrome oxidase, α-ketoglutarate dehydrogenase, and thioredoxin-dependent peroxide reductase after ischemia in KO but not in WT animals. PKC-ε deletion prevented ischemia-induced increases in oxidant production. Plasma creatinine levels increased 12-fold in WT and 3-fold in KO ischemic mice. PKC-ε deletion reduced tubular necrosis, brush border loss, and distal segment damage in ischemic kidneys. PKC-ε activation in hypoxic RPTC in primary culture exacerbated, whereas PKC-ε inhibition reduced, decreases in: 1) complex I- and complex II-coupled state 3 respirations and 2) activities of complexes I, III, and IV. We conclude that PKC-ε activation mediates 1) dysfunction of complexes I and III of the respiratory chain, 2) oxidant production, 3) morphological damage to the kidney, and 4) decreases in renal functions after ischemia.
Keywords: acute kidney injury; electron transport chain; hypoxia; ischemia and reperfusion; mitochondria; proteomics; renal proximal tubular cells.
Copyright © 2017 the American Physiological Society.
Figures
Comment in
-
Acute kidney injury: Loss of PKC-ɛ protects against IRI.Nat Rev Nephrol. 2016 Dec;12(12):714. doi: 10.1038/nrneph.2016.162. Epub 2016 Nov 7. Nat Rev Nephrol. 2016. PMID: 27818505 No abstract available.
References
-
- Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, Bolli R, Ping P. Mitochondrial PKCε and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCε-MAPK interactions and differential MAPK activation in PKCε-induced cardioprotection. Circ Res 90: 390–397, 2002. - PubMed
-
- Bak S, León I, Jensen O, Højlund K. Tissue specific phosphorylation of mitochondrial proteins isolated from rat liver, heart muscle, and skeletal muscle. J Proteome Res 12: 4327–4339, 2013. - PubMed
-
- Bender E, Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett 466: 130–134, 2000. - PubMed
-
- Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 14: 2199–2210, 2003. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials