

Metagenomic survey of methanesulfonic acid (MSA) catabolic genes in an Atlantic Ocean surface water sample and in a partial enrichment
- PMID: 27761315
- PMCID: PMC5068391
- DOI: 10.7717/peerj.2498
Metagenomic survey of methanesulfonic acid (MSA) catabolic genes in an Atlantic Ocean surface water sample and in a partial enrichment
Abstract
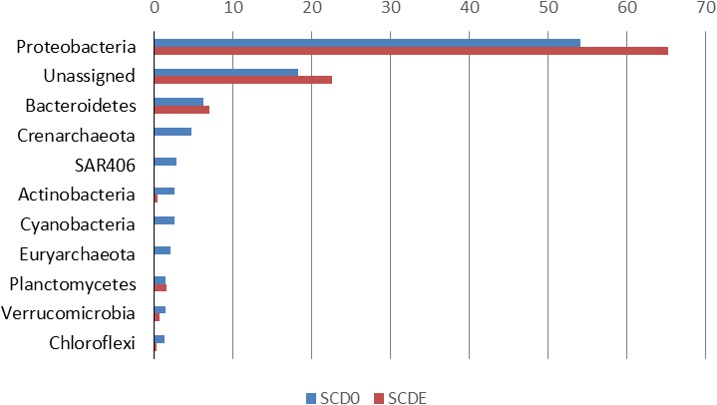
Methanesulfonic acid (MSA) is a relevant intermediate of the biogeochemical cycle of sulfur and environmental microorganisms assume an important role in the mineralization of this compound. Several methylotrophic bacterial strains able to grow on MSA have been isolated from soil or marine water and two conserved operons, msmABCD coding for MSA monooxygenase and msmEFGH coding for a transport system, have been repeatedly encountered in most of these strains. Homologous sequences have also been amplified directly from the environment or observed in marine metagenomic data, but these showed a base composition (G + C content) very different from their counterparts from cultivated bacteria. The aim of this study was to understand which microorganisms within the coastal surface oceanic microflora responded to MSA as a nutrient and how the community evolved in the early phases of an enrichment by means of metagenome and gene-targeted amplicon sequencing. From the phylogenetic point of view, the community shifted significantly with the disappearance of all signals related to the Archaea, the Pelagibacteraceae and phylum SAR406, and the increase in methylotroph-harboring taxa, accompanied by other groups so far not known to comprise methylotrophs such as the Hyphomonadaceae. At the functional level, the abundance of several genes related to sulfur metabolism and methylotrophy increased during the enrichment and the allelic distribution of gene msmA diagnostic for MSA monooxygenase altered considerably. Even more dramatic was the disappearance of MSA import-related gene msmE, which suggests that alternative transporters must be present in the enriched community and illustrate the inadequacy of msmE as an ecofunctional marker for MSA degradation at sea.
Keywords: Bacteria; Biogeochemical cycle; Gene; Metagenomics; Methanesulfonic acid; Ocean; Sulfur.
Conflict of interest statement
The authors declare there are no competing interests.
Figures







Similar articles
-
Methanesulfonate (MSA) Catabolic Genes from Marine and Estuarine Bacteria.PLoS One. 2015 May 15;10(5):e0125735. doi: 10.1371/journal.pone.0125735. eCollection 2015. PLoS One. 2015. PMID: 25978049 Free PMC article.
-
Identification, mutagenesis, and transcriptional analysis of the methanesulfonate transport operon of Methylosulfonomonas methylovora.Appl Environ Microbiol. 2006 Jan;72(1):276-83. doi: 10.1128/AEM.72.1.276-283.2006. Appl Environ Microbiol. 2006. PMID: 16391054 Free PMC article.
-
Duplicate copies of genes encoding methanesulfonate monooxygenase in Marinosulfonomonas methylotropha strain TR3 and detection of methanesulfonate utilizers in the environment.Appl Environ Microbiol. 2002 Jan;68(1):289-96. doi: 10.1128/AEM.68.1.289-296.2002. Appl Environ Microbiol. 2002. PMID: 11772638 Free PMC article.
-
Evidence of methanesulfonate utilizers in the Sargasso Sea metagenome.J Basic Microbiol. 2009 Sep;49 Suppl 1:S24-30. doi: 10.1002/jobm.200800223. J Basic Microbiol. 2009. PMID: 19322831
-
Methylotrophs in natural habitats: current insights through metagenomics.Appl Microbiol Biotechnol. 2015 Jul;99(14):5763-79. doi: 10.1007/s00253-015-6713-z. Epub 2015 Jun 9. Appl Microbiol Biotechnol. 2015. PMID: 26051673 Review.
References
-
- Andreae MO. The ocean as a source of atmospheric sulfur compounds. In: Buat-Ménard P, editor. The role of air-sea exchange in geochemical cycling. Dordrecht: Springer; 1986. pp. 331–362. (NATO ASI series 185).
-
- Azam F. Microbial control of oceanic carbon flux: the plot thickens. Science. 1998;280:694–696. doi: 10.1126/science.280.5364.694. - DOI
-
- Baxter NJ, Scanlan J, De Marco P, Wood AP, Murrell JC. Duplicate copies of genes encoding methanesulfonate monooxygenase in Marinosulfonomonas methylotropha strain TR3 and detection of methanesulfonate utilizers in the environment. Applied and Environmental Microbiology. 2002;68:289–296. doi: 10.1128/AEM.68.1.289. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
