

Mutant BRAF Upregulates MCL-1 to Confer Apoptosis Resistance that Is Reversed by MCL-1 Antagonism and Cobimetinib in Colorectal Cancer
- PMID: 27765849
- PMCID: PMC5136313
- DOI: 10.1158/1535-7163.MCT-16-0017
Mutant BRAF Upregulates MCL-1 to Confer Apoptosis Resistance that Is Reversed by MCL-1 Antagonism and Cobimetinib in Colorectal Cancer
Abstract
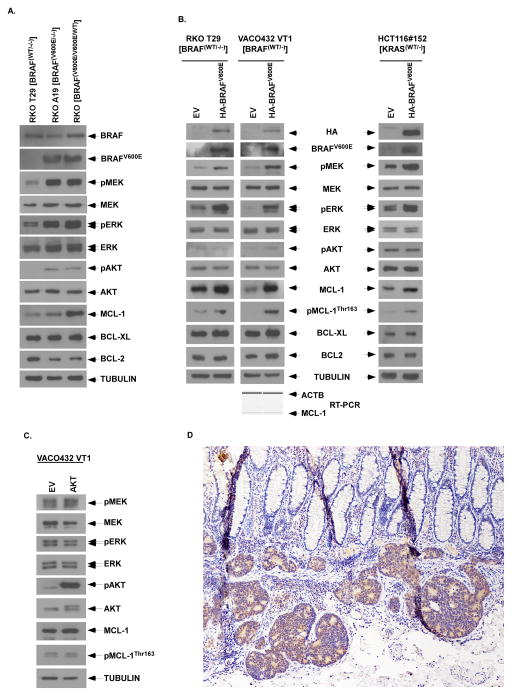
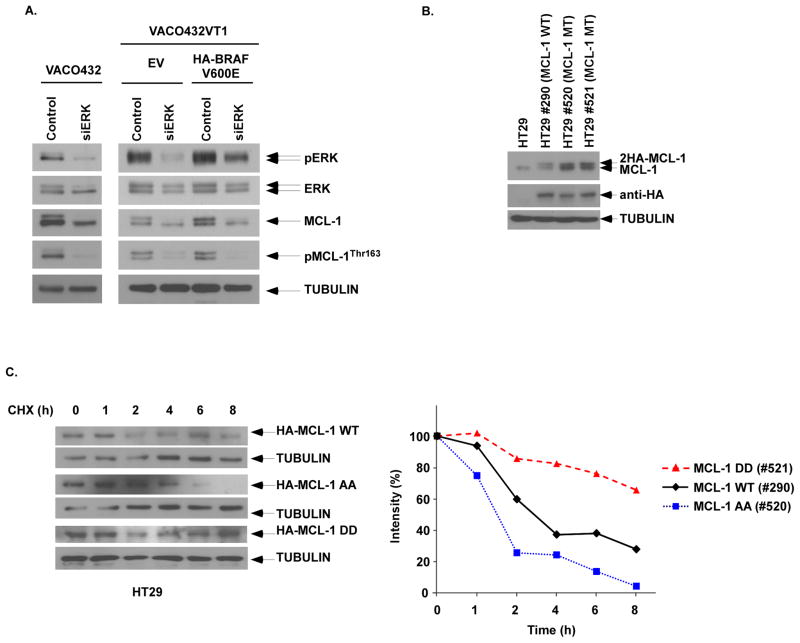
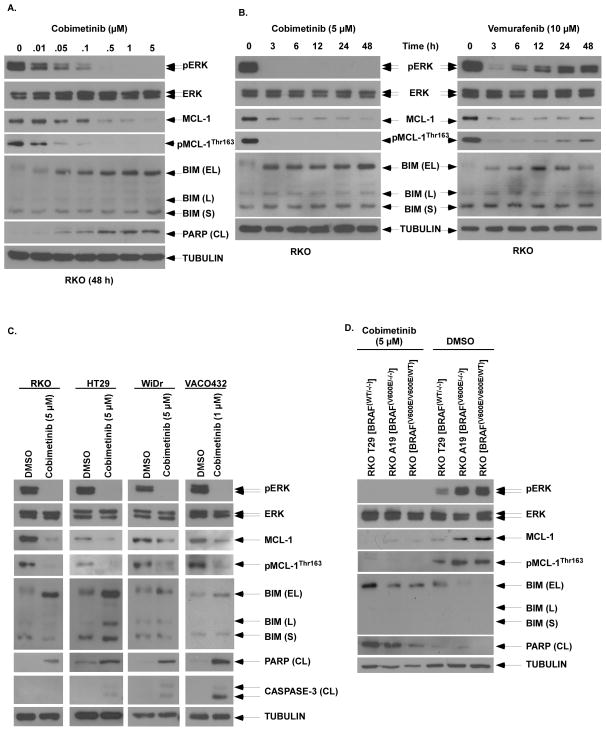
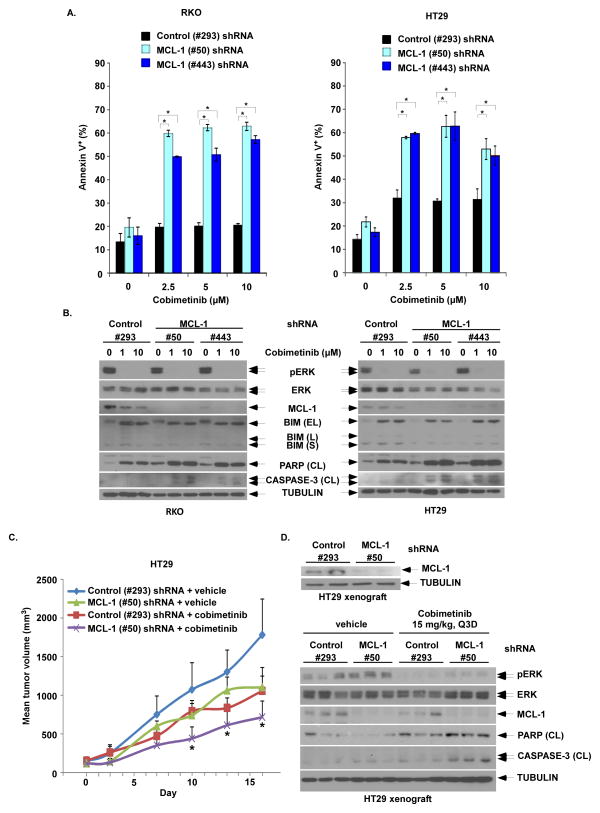
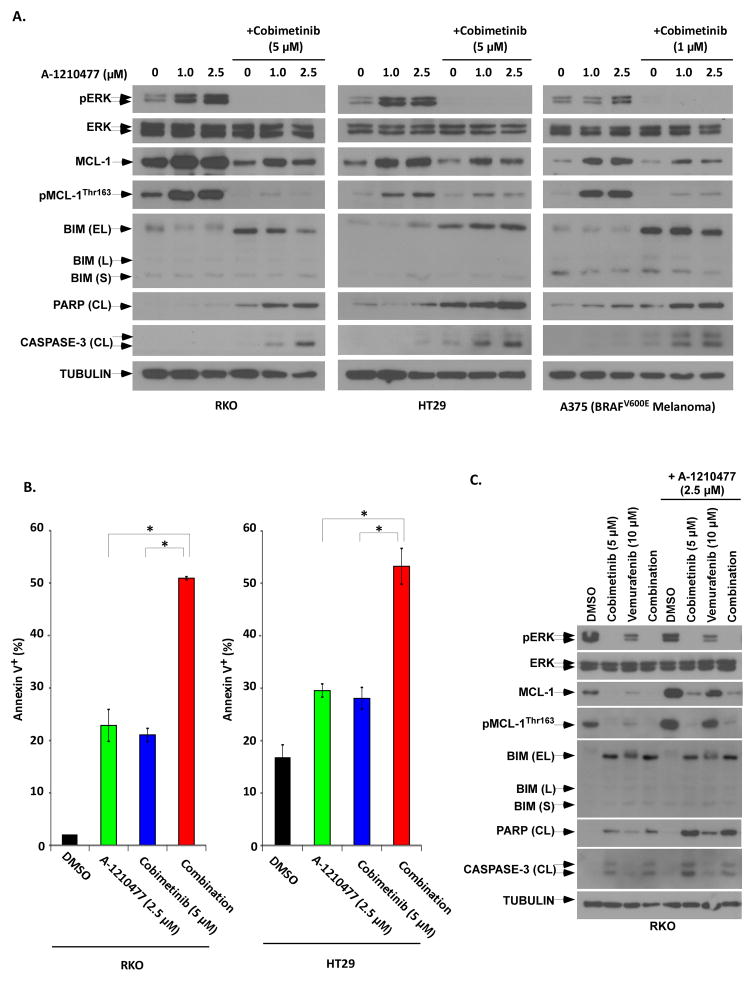
Oncogenic BRAFV600E mutations activate MAPK signaling and are associated with treatment resistance and poor prognosis in patients with colorectal cancer. In BRAFV600E-mutant colorectal cancers, treatment failure may be related to BRAFV600E-mediated apoptosis resistance that occurs by an as yet undefined mechanism. We found that BRAFV600E can upregulate anti-apoptotic MCL-1 in a gene dose-dependent manner using colorectal cancer cell lines isogenic for BRAF BRAFV600E-induced MCL-1 upregulation was confirmed by ectopic BRAFV600E expression that activated MEK/ERK signaling to phosphorylate (MCL-1Thr163) and stabilize MCL-1. Upregulation of MCL-1 was mediated by MEK/ERK shown by the ability of ERK siRNA to suppress MCL-1. Stabilization of MCL-1 by phosphorylation was shown by a phosphorylation-mimicking mutant and an unphosphorylated MCL-1 mutant that decreased or increased MCL-1 protein turnover, respectively. MEK/ERK inhibition by cobimetinib suppressed MCL-1 expression/phosphorylation and induced proapoptotic BIM to a greater extent than did vemurafenib in BRAFV600E cell lines. MCL-1 knockdown versus control shRNA significantly enhanced cobimetinib-induced apoptosis in vitro and in HT29 colon cancer xenografts. The small-molecule MCL-1 inhibitor, A-1210477, also enhanced cobimetinib-induced apoptosis in vitro that was due to disruption of the interaction of MCL-1 with proapoptotic BAK and BIM. Knockdown of BIM attenuated BAX, but not BAK, activation by cobimetinib plus A-1210477. In summary, BRAFV600E-mediated MEK/ERK activation can upregulate MCL-1 by phosphorylation/stabilization to confer apoptosis resistance that can be reversed by MCL-1 antagonism combined with cobimetinib, suggesting a novel therapeutic strategy against BRAFV600E-mutant CRCs. Mol Cancer Ther; 15(12); 3015-27. ©2016 AACR.
©2016 American Association for Cancer Research.
Conflict of interest statement
Conflict of Interest. The authors report no conflicts related to the content of the manuscript.
Figures

References
-
- Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501:232–6. - PubMed
-
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. - PubMed
-
- Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466–74. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
