

Characterization of Pharmacologic and Pharmacokinetic Properties of CCX168, a Potent and Selective Orally Administered Complement 5a Receptor Inhibitor, Based on Preclinical Evaluation and Randomized Phase 1 Clinical Study
- PMID: 27768695
- PMCID: PMC5074546
- DOI: 10.1371/journal.pone.0164646
Characterization of Pharmacologic and Pharmacokinetic Properties of CCX168, a Potent and Selective Orally Administered Complement 5a Receptor Inhibitor, Based on Preclinical Evaluation and Randomized Phase 1 Clinical Study
Erratum in
-
Correction: Characterization of Pharmacologic and Pharmacokinetic Properties of CCX168, a Potent and Selective Orally Administered Complement 5a Receptor Inhibitor, Based on Preclinical Evaluation and Randomized Phase 1 Clinical Study.PLoS One. 2019 Jan 4;14(1):e0210593. doi: 10.1371/journal.pone.0210593. eCollection 2019. PLoS One. 2019. PMID: 30608991 Free PMC article.
Abstract
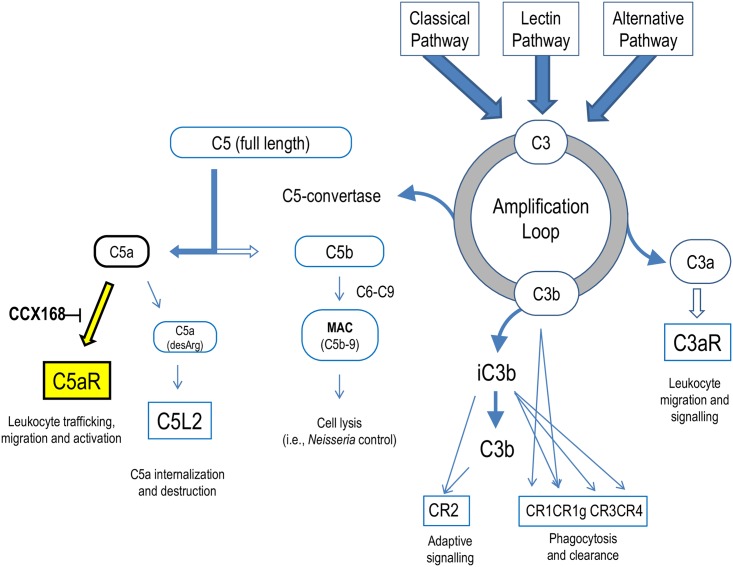
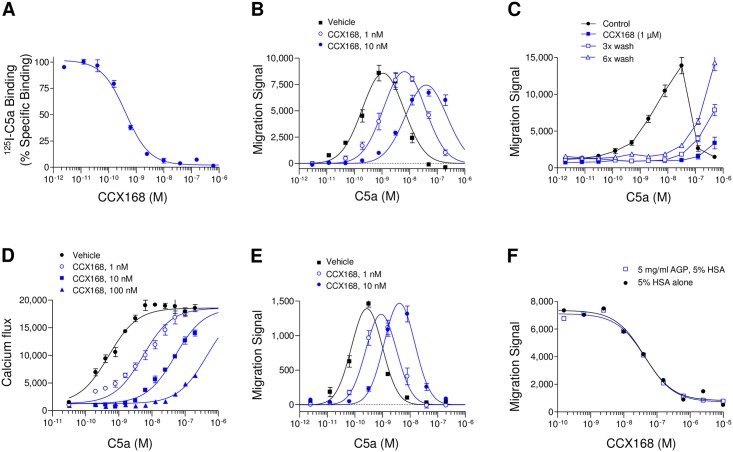
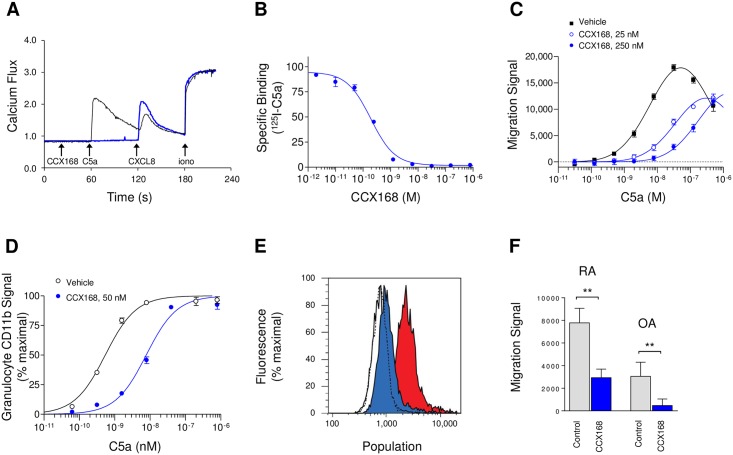
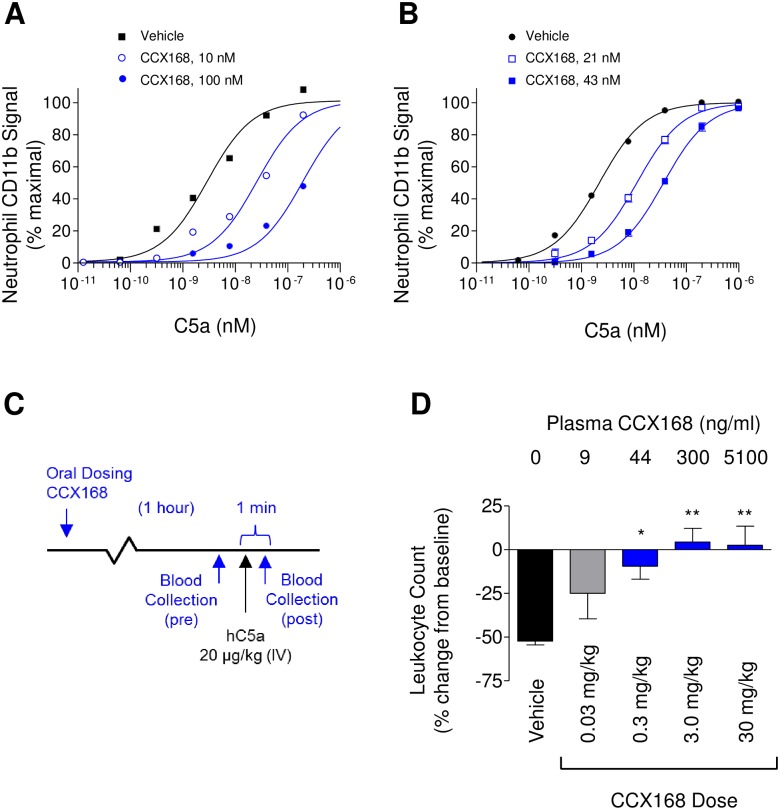
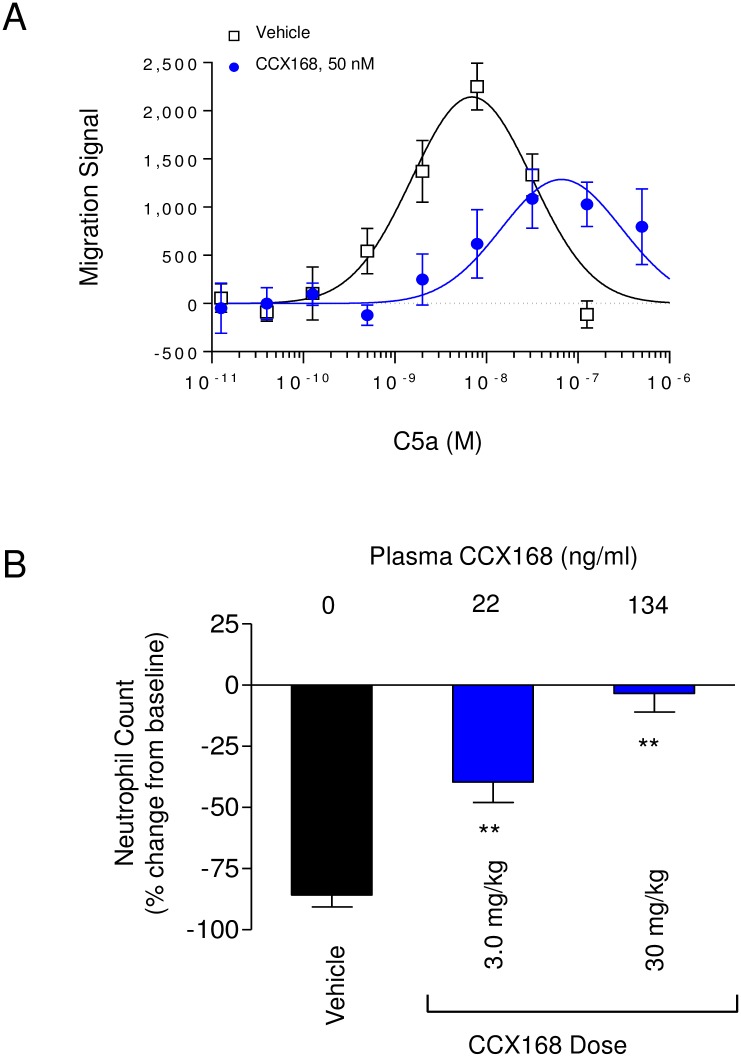
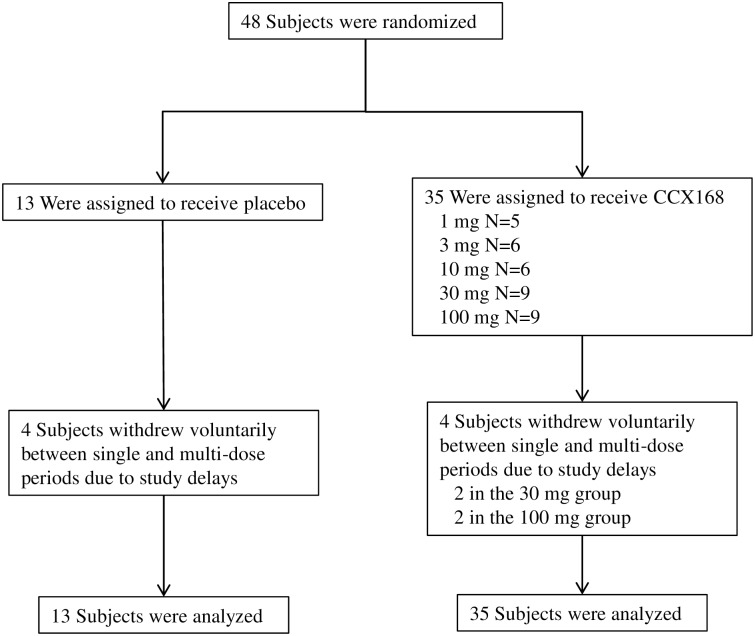
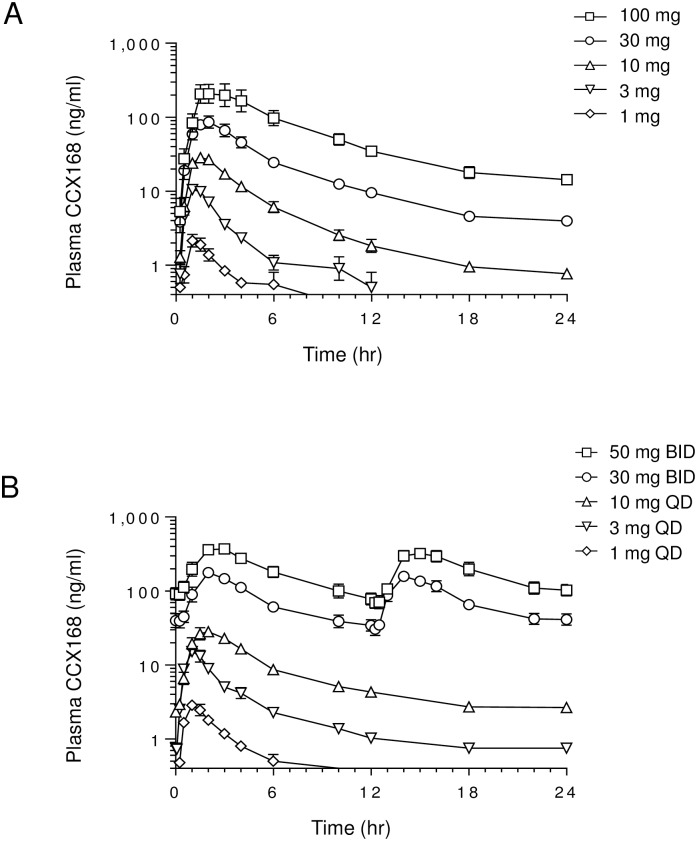
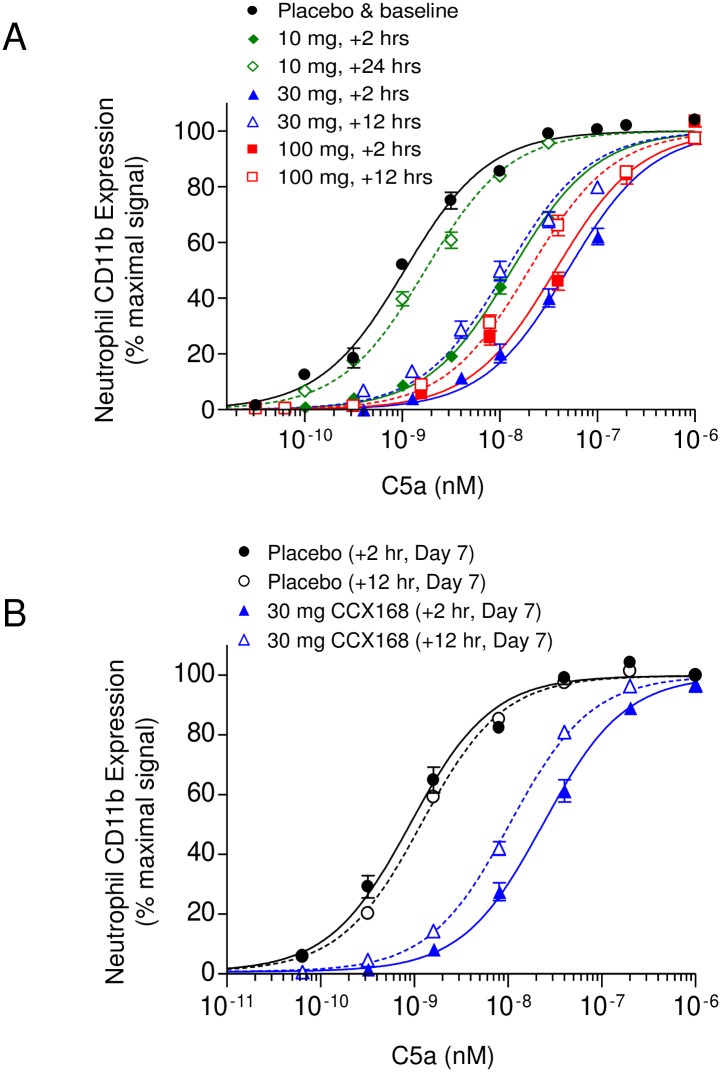
The complement 5a receptor has been an attractive therapeutic target for many autoimmune and inflammatory disorders. However, development of a selective and potent C5aR antagonist has been challenging. Here we describe the characterization of CCX168 (avacopan), an orally administered selective and potent C5aR inhibitor. CCX168 blocked the C5a binding, C5a-mediated migration, calcium mobilization, and CD11b upregulation in U937 cells as well as in freshly isolated human neutrophils. CCX168 retains high potency when present in human blood. A transgenic human C5aR knock-in mouse model allowed comparison of the in vitro and in vivo efficacy of the molecule. CCX168 effectively blocked migration in in vitro and ex vivo chemotaxis assays, and it blocked the C5a-mediated neutrophil vascular endothelial margination. CCX168 was effective in migration and neutrophil margination assays in cynomolgus monkeys. This thorough in vitro and preclinical characterization enabled progression of CCX168 into the clinic and testing of its safety, tolerability, pharmacokinetic, and pharmacodynamic profiles in a Phase 1 clinical trial in 48 healthy volunteers. CCX168 was shown to be well tolerated across a broad dose range (1 to 100 mg) and it showed dose-dependent pharmacokinetics. An oral dose of 30 mg CCX168 given twice daily blocked the C5a-induced upregulation of CD11b in circulating neutrophils by 94% or greater throughout the entire day, demonstrating essentially complete target coverage. This dose regimen is being tested in clinical trials in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Trial Registration ISRCTN registry with trial ID ISRCTN13564773.
Conflict of interest statement
The work was funded by ChemoCentryx, Inc. The ChemoCentryx, Inc. website notes that CCX168 is under development by this company and have market exclusivity. This does not alter our adherence to PLOS ONE policies on sharing data and materials.
Figures
References
-
- Speth C, Prodinger W, Wurzner R, Stoiber H, Dierich M. Complement In: Paul WE, editor. Fundamental Immunology. Sixth ed Philadelphia, PA: Lippincott Williams & Wilkins; 2008. pp. 1047–1078.
-
- Sahu A, Lambris JD. Complement inhibitors: a resurgent concept in anti-inflammatory therapeutics. Immunopharmacol. 2000;49:133–148. - PubMed
-
- Powers JP, Dairaghi DJ, Jaen JC. Advances in the discovery of C5a receptor antagonists. Ann Reports Med Chem. 2011;46:171–186.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
