

Protein folding, binding, and droplet formation in cell-like conditions
- PMID: 27771543
- PMCID: PMC5397379
- DOI: 10.1016/j.sbi.2016.10.006
Protein folding, binding, and droplet formation in cell-like conditions
Abstract
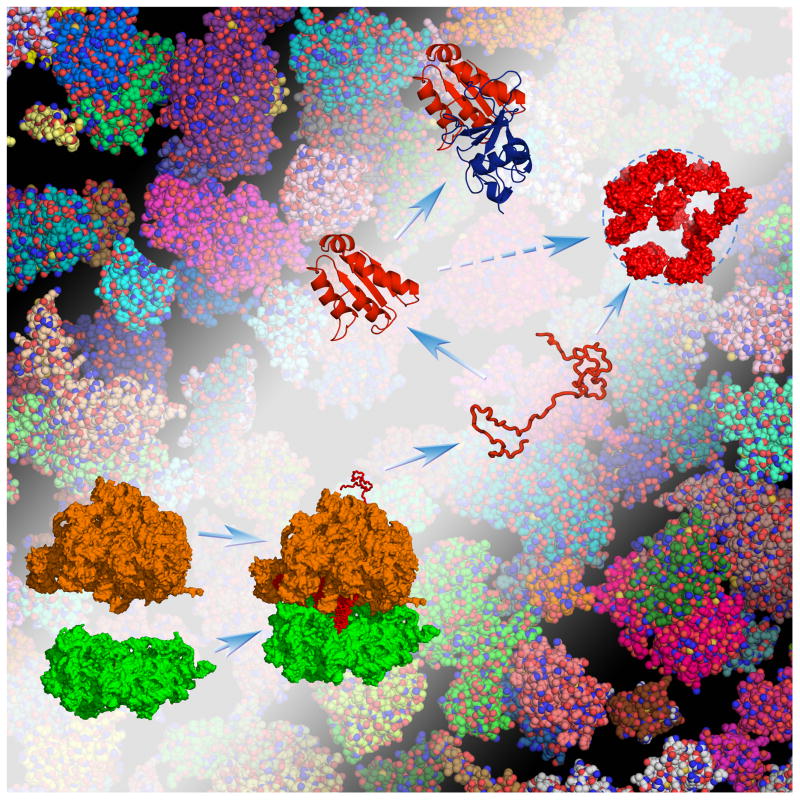
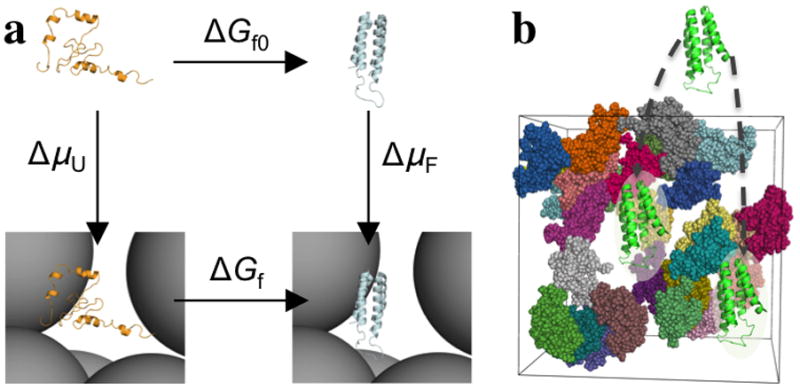
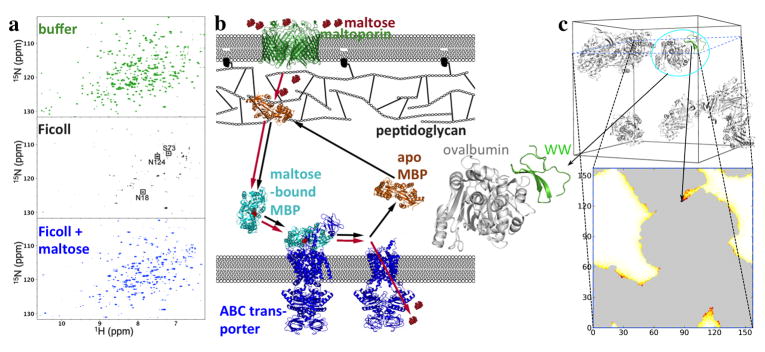
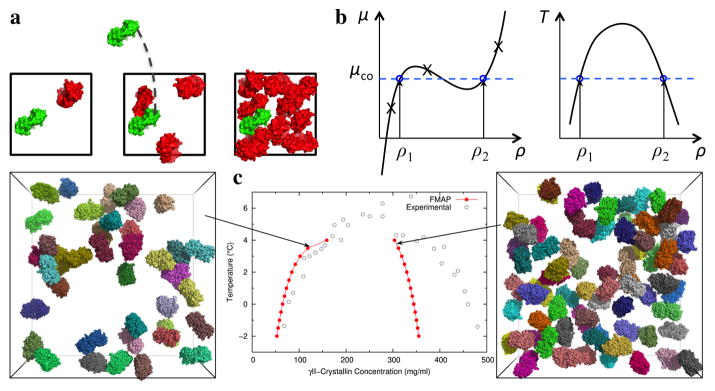
The many bystander macromolecules in the crowded cellular environments present both steric repulsion and weak attraction to proteins undergoing folding or binding and hence impact the thermodynamic and kinetic properties of these processes. The weak but nonrandom binding with bystander macromolecules may facilitate subcellular localization and biological function. Weak binding also leads to the emergence of a protein-rich droplet phase, which has been implicated in regulating a variety of cellular functions. All these important problems can now be addressed by realistic modeling of intermolecular interactions. Configurational sampling of concentrated protein solutions is an ongoing challenge.
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Effects of macromolecular crowding on protein folding and aggregation studied by density functional theory: dynamics.Phys Rev E Stat Nonlin Soft Matter Phys. 2002 Nov;66(5 Pt 1):051902. doi: 10.1103/PhysRevE.66.051902. Epub 2002 Nov 7. Phys Rev E Stat Nonlin Soft Matter Phys. 2002. PMID: 12513518
-
NMR studies of protein folding and binding in cells and cell-like environments.Curr Opin Struct Biol. 2015 Feb;30:7-16. doi: 10.1016/j.sbi.2014.10.004. Epub 2014 Dec 3. Curr Opin Struct Biol. 2015. PMID: 25479354 Review.
-
Topology-based modeling of intrinsically disordered proteins: balancing intrinsic folding and intermolecular interactions.Proteins. 2011 Apr;79(4):1251-66. doi: 10.1002/prot.22960. Epub 2011 Jan 25. Proteins. 2011. PMID: 21268115
-
Dynamics and mechanisms of coupled protein folding and binding reactions.Curr Opin Struct Biol. 2012 Feb;22(1):21-9. doi: 10.1016/j.sbi.2011.09.010. Epub 2011 Nov 29. Curr Opin Struct Biol. 2012. PMID: 22129832 Review.
-
Uncoupled binding and folding of immune signaling-related intrinsically disordered proteins.Prog Biophys Mol Biol. 2011 Sep;106(3):525-36. doi: 10.1016/j.pbiomolbio.2011.08.005. Epub 2011 Aug 18. Prog Biophys Mol Biol. 2011. PMID: 21867726 Review.
Cited by
-
Protein Association in Solution: Statistical Mechanical Modeling.Biomolecules. 2023 Nov 24;13(12):1703. doi: 10.3390/biom13121703. Biomolecules. 2023. PMID: 38136574 Free PMC article. Review.
-
Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation.Chem Rev. 2018 Feb 28;118(4):1691-1741. doi: 10.1021/acs.chemrev.7b00305. Epub 2018 Jan 10. Chem Rev. 2018. PMID: 29319301 Free PMC article. Review.
-
Charge-driven condensation of RNA and proteins suggests broad role of phase separation in cytoplasmic environments.Elife. 2021 Jan 26;10:e64004. doi: 10.7554/eLife.64004. Elife. 2021. PMID: 33496264 Free PMC article.
-
Both Ligands and Macromolecular Crowders Preferentially Bind to Closed Conformations of Maltose Binding Protein.Biochemistry. 2019 Apr 30;58(17):2208-2217. doi: 10.1021/acs.biochem.9b00154. Epub 2019 Apr 12. Biochemistry. 2019. PMID: 30950267 Free PMC article.
-
Liquid-Liquid Phase Separation of Patchy Particles Illuminates Diverse Effects of Regulatory Components on Protein Droplet Formation.Sci Rep. 2018 Apr 30;8(1):6728. doi: 10.1038/s41598-018-25132-1. Sci Rep. 2018. PMID: 29712961 Free PMC article.
References
-
- Minton AP, Wilf J. Effect of macromolecular crowding upon the structure and function of an enzyme: glyceraldehyde-3-phosphate dehydrogenase. Biochemistry. 1981;20:4821–4826. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
