

Marked Differences in C9orf72 Methylation Status and Isoform Expression between C9/ALS Human Embryonic and Induced Pluripotent Stem Cells
- PMID: 27773700
- PMCID: PMC5106522
- DOI: 10.1016/j.stemcr.2016.09.011
Marked Differences in C9orf72 Methylation Status and Isoform Expression between C9/ALS Human Embryonic and Induced Pluripotent Stem Cells
Abstract
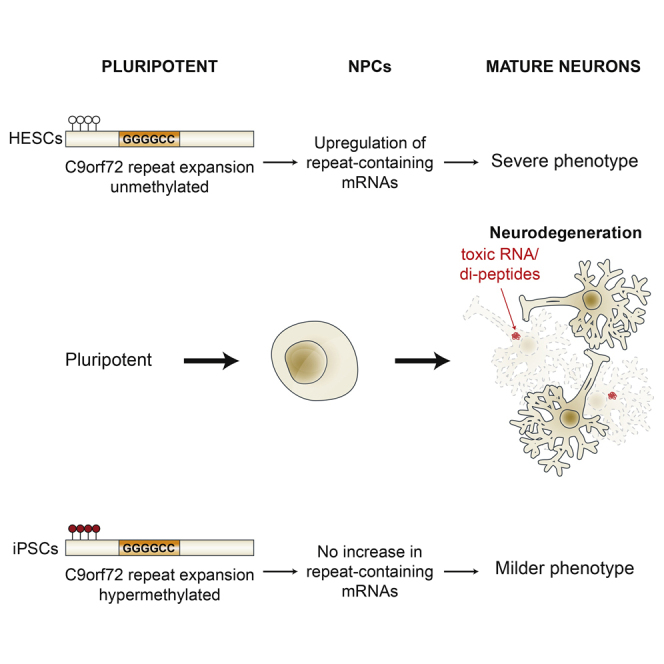
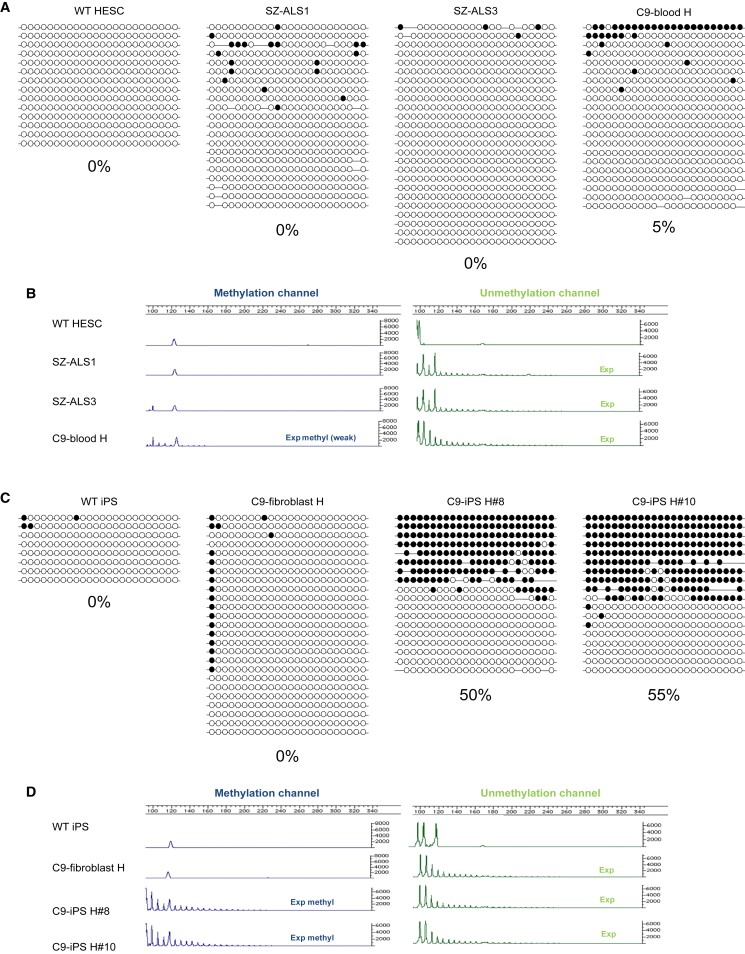
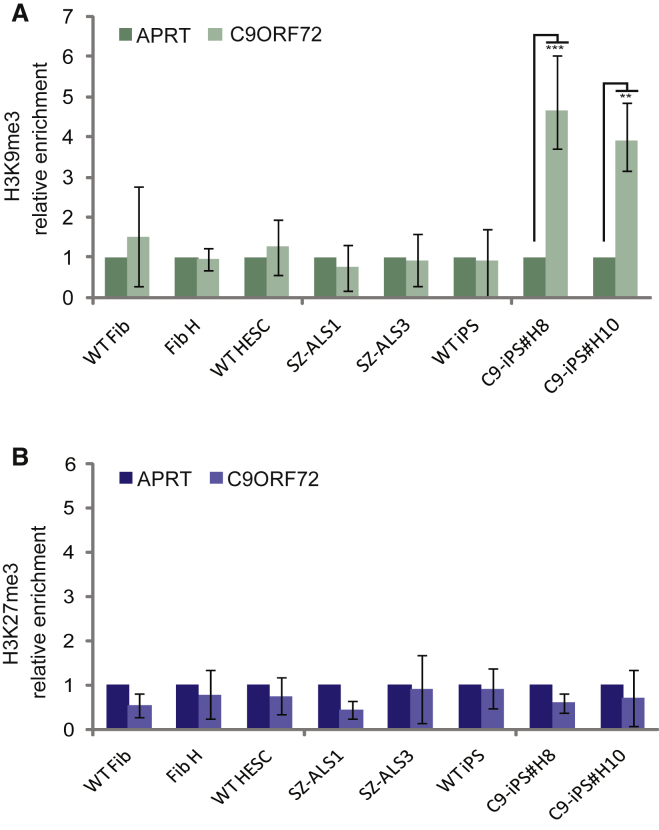
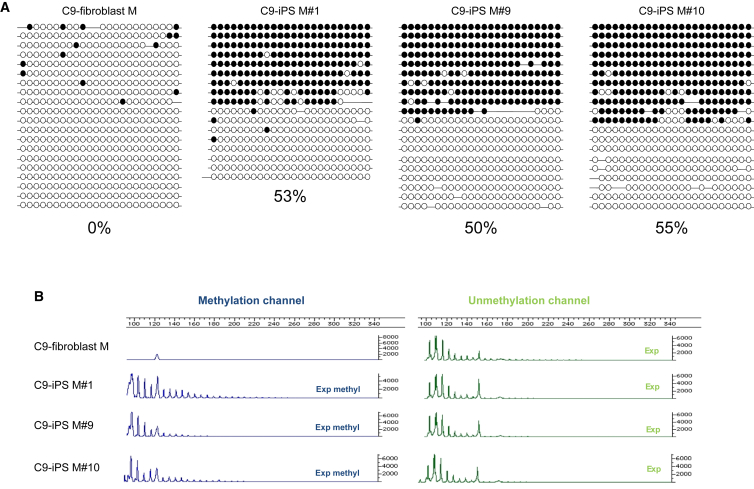
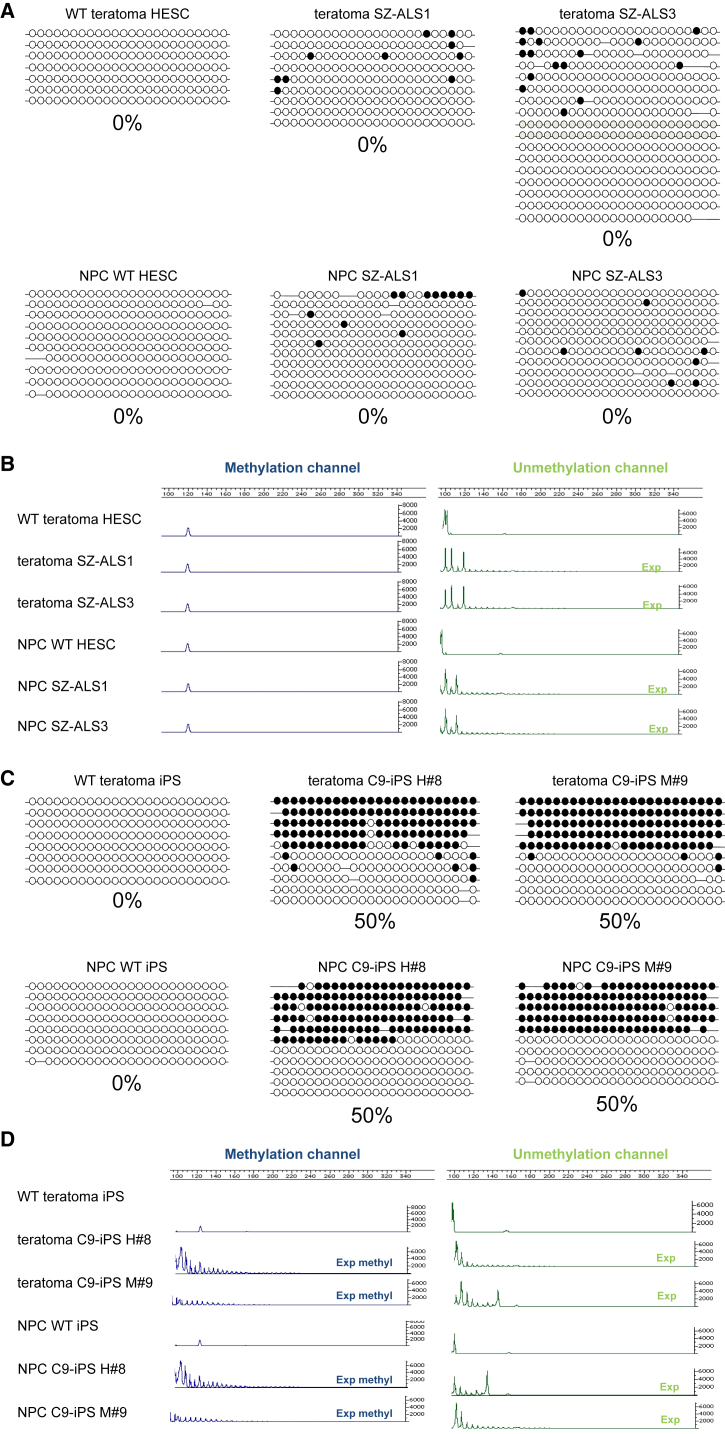
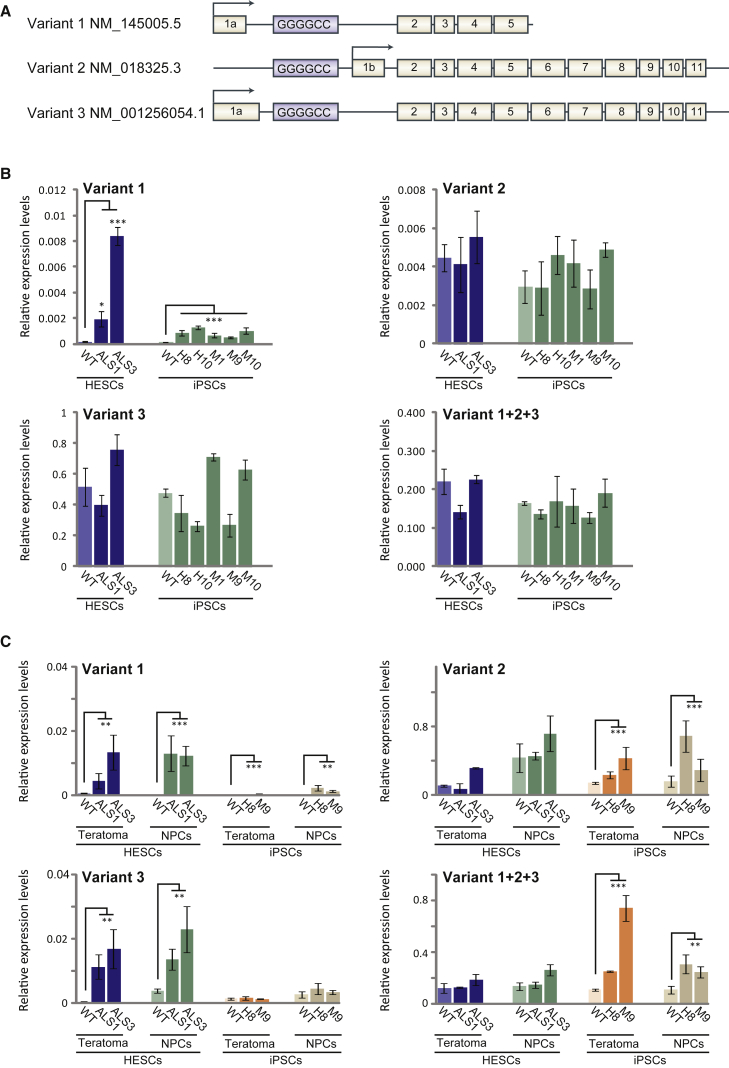
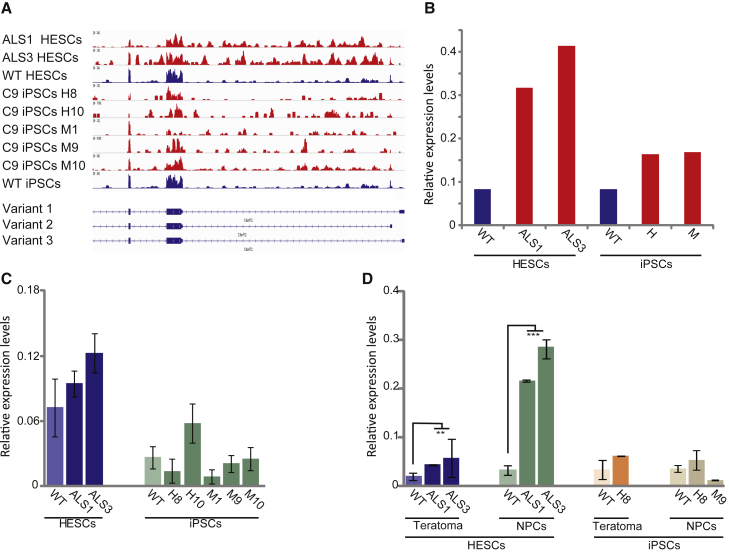
We established two human embryonic stem cell (hESC) lines with a GGGGCC expansion in the C9orf72 gene (C9), and compared them with haploidentical and unrelated C9 induced pluripotent stem cells (iPSCs). We found a marked difference in C9 methylation between the cells. hESCs and parental fibroblasts are entirely unmethylated while the iPSCs are hypermethylated. In addition, we show that the expansion alters promoter usage and interferes with the proper splicing of intron 1, eventually leading to the accumulation of repeat-containing mRNA following neural differentiation. These changes are attenuated in C9 iPSCs, presumably owing to hypermethylation. Altogether, this study highlights the importance of neural differentiation in the pathogenesis of disease and points to the potential role of hypermethylation as a neuroprotective mechanism against pathogenic mRNAs, envisaging a milder phenotype in C9 iPSCs.
Keywords: C9/ALS; CpG islands; DNA methylation; disease modelling; neurodegeneration; pluripotent stem cells; unstable repeat expansions.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Almeida S., Gascon E., Tran H., Chou H.J., Gendron T.F., Degroot S., Tapper A.R., Sellier C., Charlet-Berguerand N., Karydas A. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126:385–399. - PMC - PubMed
-
- Bauer P.O. Methylation of C9orf72 expansion reduces RNA foci formation and dipeptide-repeat proteins expression in cells. Neurosci. Lett. 2016;612:204–209. - PubMed
-
- Belzil V.V., Bauer P.O., Prudencio M., Gendron T.F., Stetler C.T., Yan I.K., Pregent L., Daughrity L., Baker M.C., Rademakers R. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126:895–905. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
