

MERS-CoV recombination: implications about the reservoir and potential for adaptation
- PMID: 27774293
- PMCID: PMC4989901
- DOI: 10.1093/ve/vev023
MERS-CoV recombination: implications about the reservoir and potential for adaptation
Abstract
Recombination is a process that unlinks neighboring loci allowing for independent evolutionary trajectories within genomes of many organisms. If not properly accounted for, recombination can compromise many evolutionary analyses. In addition, when dealing with organisms that are not obligately sexually reproducing, recombination gives insight into the rate at which distinct genetic lineages come into contact. Since June 2012, Middle East respiratory syndrome coronavirus (MERS-CoV) has caused 1,106 laboratory-confirmed infections, with 421 MERS-CoV-associated deaths as of 16 April 2015. Although bats are considered as the likely ultimate source of zoonotic betacoronaviruses, dromedary camels have been consistently implicated as the source of current human infections in the Middle East. In this article, we use phylogenetic methods and simulations to show that MERS-CoV genome has likely undergone numerous recombinations recently. Recombination in MERS-CoV implies frequent co-infection with distinct lineages of MERS-CoV, probably in camels given the current understanding of MERS-CoV epidemiology.
Keywords: MERS; MERS-CoV; co-infection; coronavirus; homoplasy; recombination.
Figures

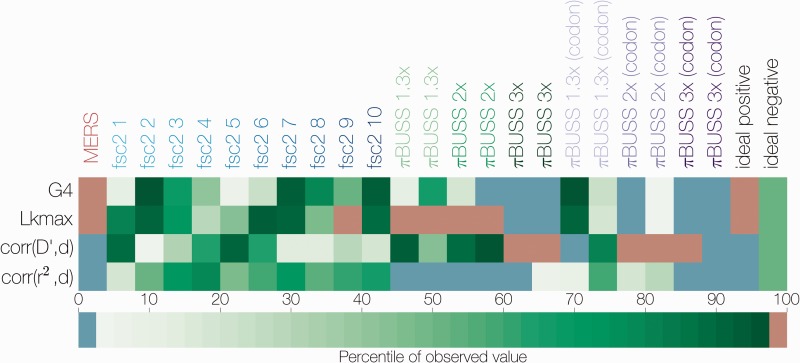
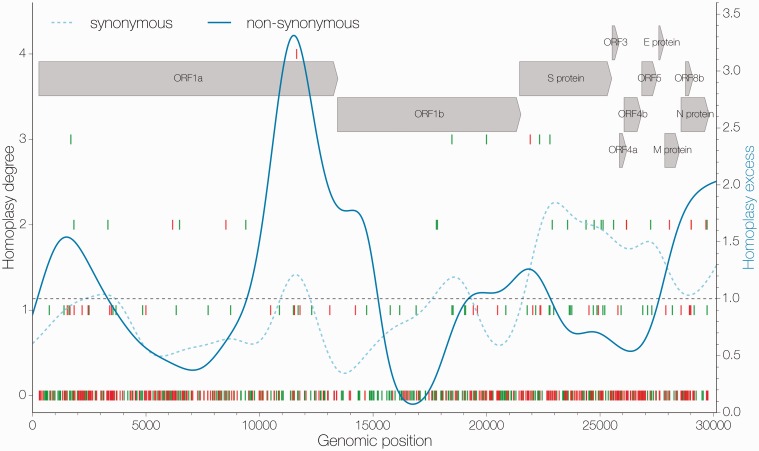



References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
