

Driving forces behind the evolution of the Aleutian mink disease parvovirus in the context of intensive farming
- PMID: 27774297
- PMCID: PMC4989880
- DOI: 10.1093/ve/vew004
Driving forces behind the evolution of the Aleutian mink disease parvovirus in the context of intensive farming
Abstract
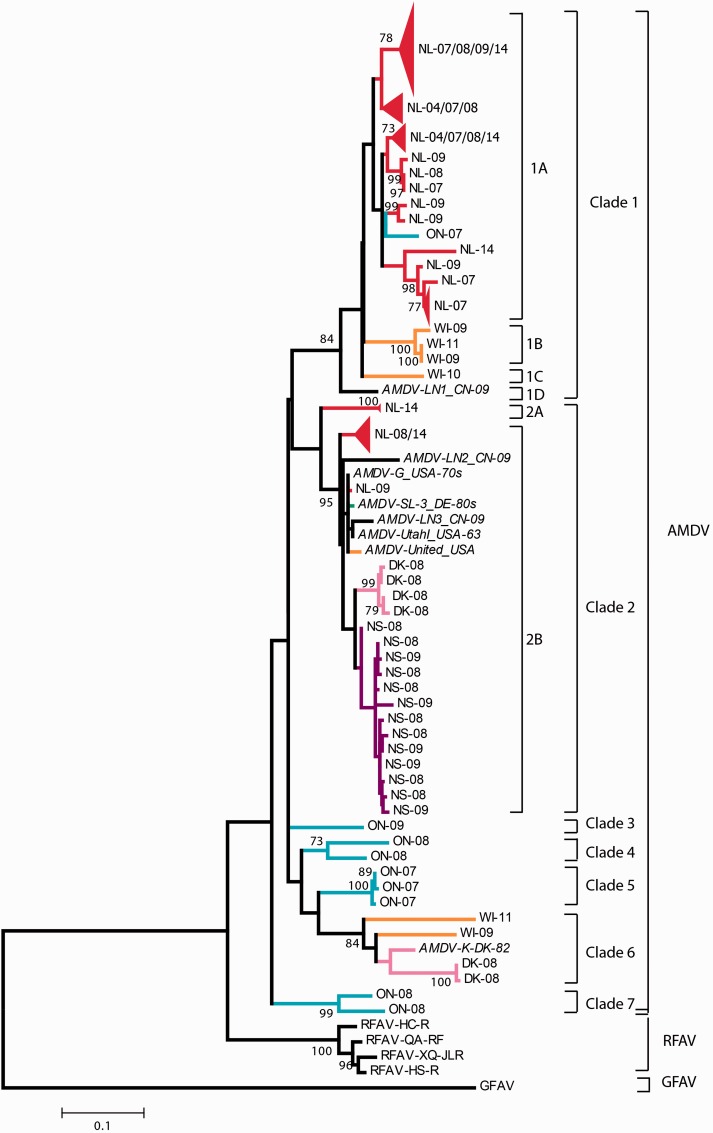
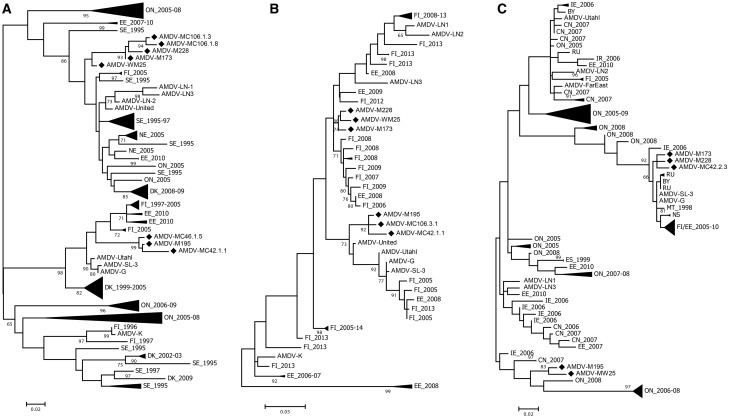
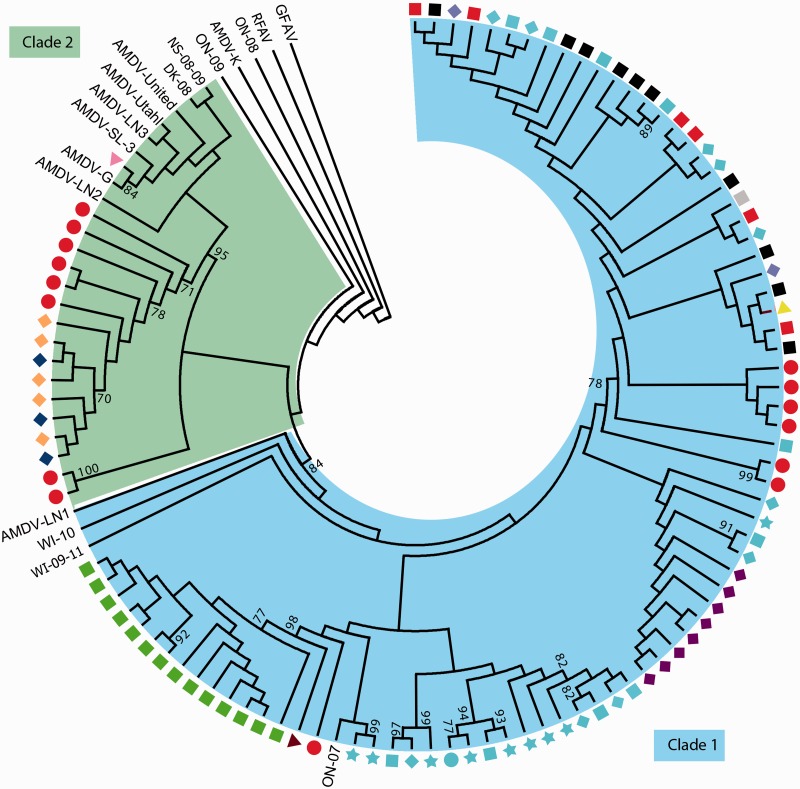
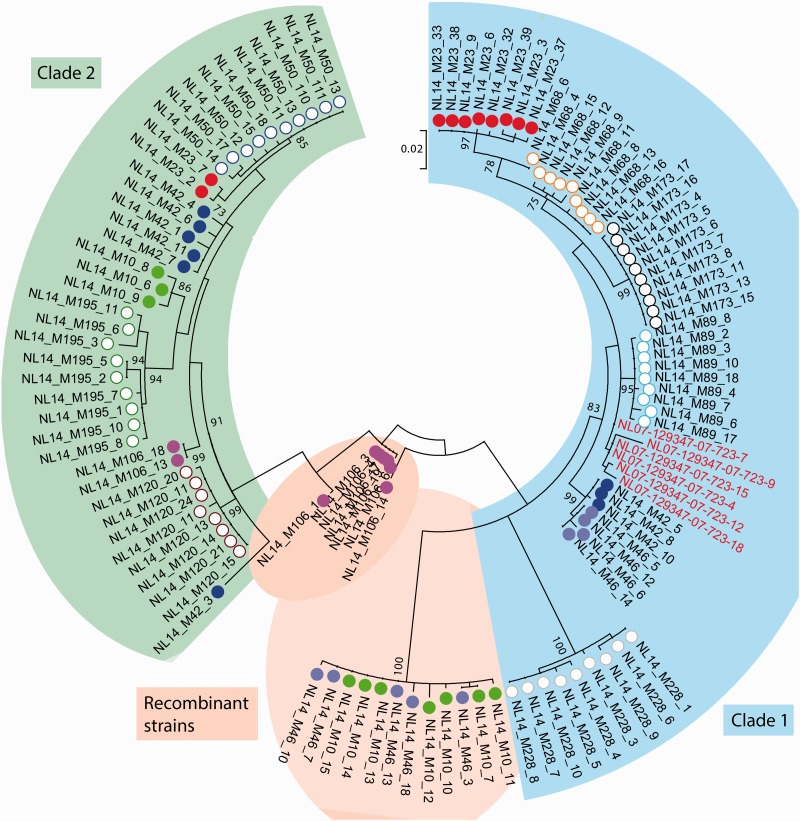
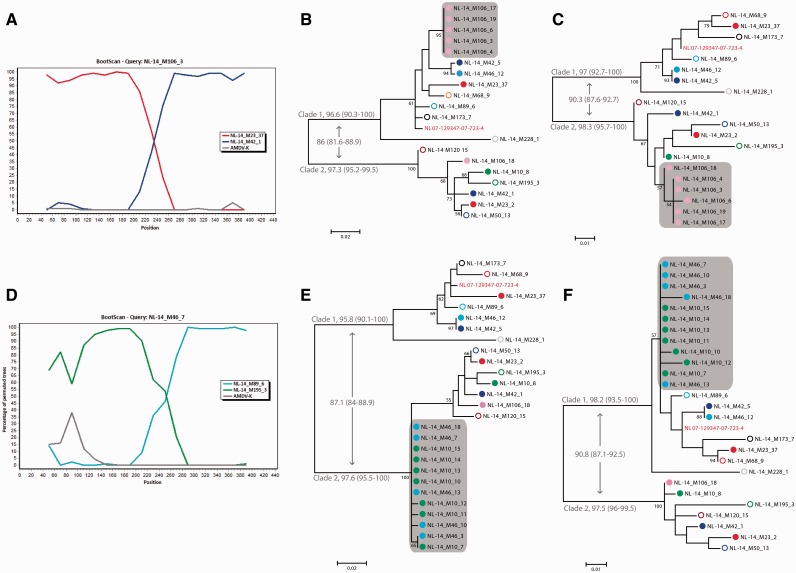
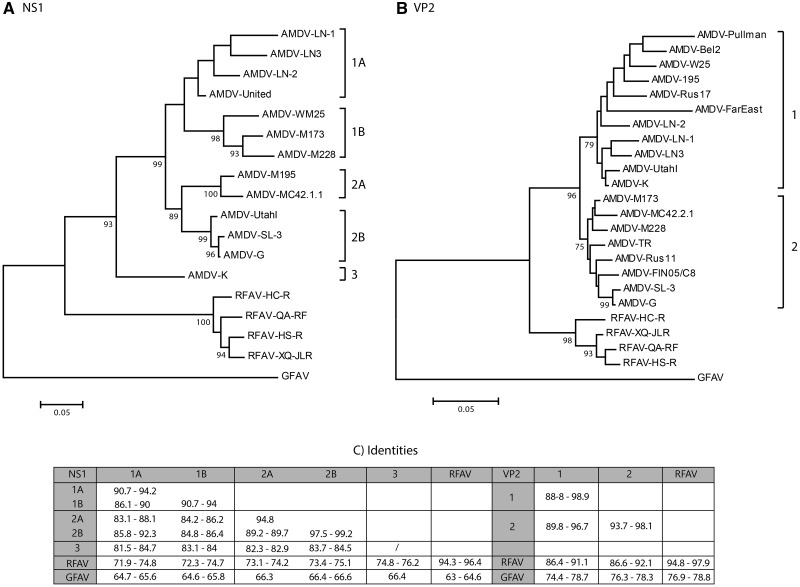
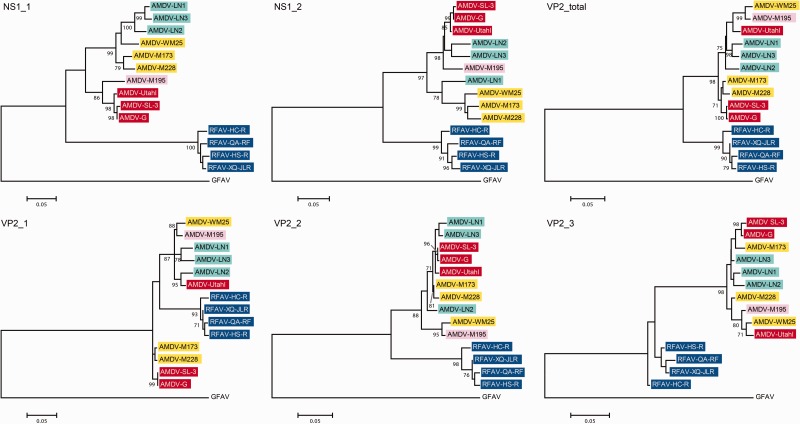
Aleutian mink disease virus (AMDV) causes plasmacytosis, an immune complex-associated syndrome that affects wild and farmed mink. The virus can also infect other small mammals (e.g., ferrets, skunks, ermines, and raccoons), but the disease in these hosts has been studied less. In 2007, a mink plasmacytosis outbreak began on the Island of Newfoundland, and the virus has been endemic in farms since then. In this study, we evaluated the molecular epidemiology of AMDV in farmed and wild animals of Newfoundland since before the beginning of the outbreak and investigated the epidemic in a global context by studying AMDV worldwide, thereby examining its diffusion and phylogeography. Furthermore, AMDV evolution was examined in the context of intensive farming, where host population dynamics strongly influence viral evolution. Partial NS1 sequences and several complete genomes were obtained from Newfoundland viruses and analyzed along with numerous sequences from other locations worldwide that were either obtained as part of this study or from public databases. We observed very high viral diversity within Newfoundland and within single farms, where high rates of co-infection, recombinant viruses and polymorphisms were observed within single infected individuals. Worldwide, we documented a partial geographic distribution of strains, where viruses from different countries co-exist within clades but form country-specific subclades. Finally, we observed the occurrence of recombination and the predominance of negative selection pressure on AMDV proteins. A surprisingly low number of immunoepitopic sites were under diversifying pressure, possibly because AMDV gains no benefit by escaping the immune response as viral entry into target cells is mediated through interactions with antibodies, which therefore contribute to cell infection. In conclusion, the high prevalence of AMDV in farms facilitates the establishment of co-infections that can favor the occurrence of recombination and enhance viral diversity. Viruses are then exchanged between different farms and countries and can be introduced into the wild, with the rapidly evolving viruses producing many parallel lineages.
Keywords: AMDV; Aleutian mink disease virus; amdoparvoviruses; parvoviruses; viral evolution; viral recombination.
Figures
Similar articles
-
Multi-host dispersal of known and novel carnivore amdoparvoviruses.Virus Evol. 2020 Dec 6;6(2):veaa072. doi: 10.1093/ve/veaa072. eCollection 2020 Jul. Virus Evol. 2020. PMID: 36158990 Free PMC article.
-
Outbreak tracking of Aleutian mink disease virus (AMDV) using partial NS1 gene sequencing.Virol J. 2017 Jun 21;14(1):119. doi: 10.1186/s12985-017-0786-5. Virol J. 2017. PMID: 28637462 Free PMC article.
-
Molecular epidemiology of Aleutian mink disease virus (AMDV) in Estonia, and a global phylogeny of AMDV.Virus Res. 2015 Mar 2;199:56-61. doi: 10.1016/j.virusres.2015.01.011. Epub 2015 Jan 20. Virus Res. 2015. PMID: 25616049
-
Seroprevalence and Molecular Epidemiology of Aleutian Disease in Various Countries during 1972-2021: A Review and Meta-Analysis.Animals (Basel). 2021 Oct 15;11(10):2975. doi: 10.3390/ani11102975. Animals (Basel). 2021. PMID: 34679996 Free PMC article. Review.
-
AMDV Vaccine: Challenges and Perspectives.Viruses. 2021 Sep 14;13(9):1833. doi: 10.3390/v13091833. Viruses. 2021. PMID: 34578415 Free PMC article. Review.
Cited by
-
Selection for Favorable Health Traits: A Potential Approach to Cope with Diseases in Farm Animals.Animals (Basel). 2020 Sep 22;10(9):1717. doi: 10.3390/ani10091717. Animals (Basel). 2020. PMID: 32971980 Free PMC article. Review.
-
Natural disease and evolution of an Amdoparvovirus endemic in striped skunks (Mephitis mephitis).Transbound Emerg Dis. 2022 Sep;69(5):e1758-e1767. doi: 10.1111/tbed.14511. Epub 2022 Mar 26. Transbound Emerg Dis. 2022. PMID: 35279956 Free PMC article.
-
A new perspective on the evolution and diversity of the genus Amdoparvovirus (family Parvoviridae) through genetic characterization, structural homology modeling, and phylogenetics.Virus Evol. 2022 Jun 17;8(1):veac056. doi: 10.1093/ve/veac056. eCollection 2022. Virus Evol. 2022. PMID: 35783582 Free PMC article.
-
Endogenous amdoparvovirus-related elements reveal insights into the biology and evolution of vertebrate parvoviruses.Virus Evol. 2018 Nov 12;4(2):vey026. doi: 10.1093/ve/vey026. eCollection 2018 Jul. Virus Evol. 2018. PMID: 30443409 Free PMC article.
-
Molecular epidemiology of Aleutian mink disease virus causing outbreaks in mink farms from Southwestern Europe: a retrospective study from 2012 to 2019.J Vet Sci. 2020 Jul;21(4):e65. doi: 10.4142/jvs.2020.21.e65. J Vet Sci. 2020. PMID: 32735101 Free PMC article.
References
-
- Alexandersen S. (1986) ‘Acute Interstitial Pneumonia in Mink Kits: Experimental Reproduction of the Disease’, Veterinary Pathology, 23: 579–88. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
