

Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen)
- PMID: 27774300
- PMCID: PMC4989882
- DOI: 10.1093/ve/vew007
Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen)
Abstract
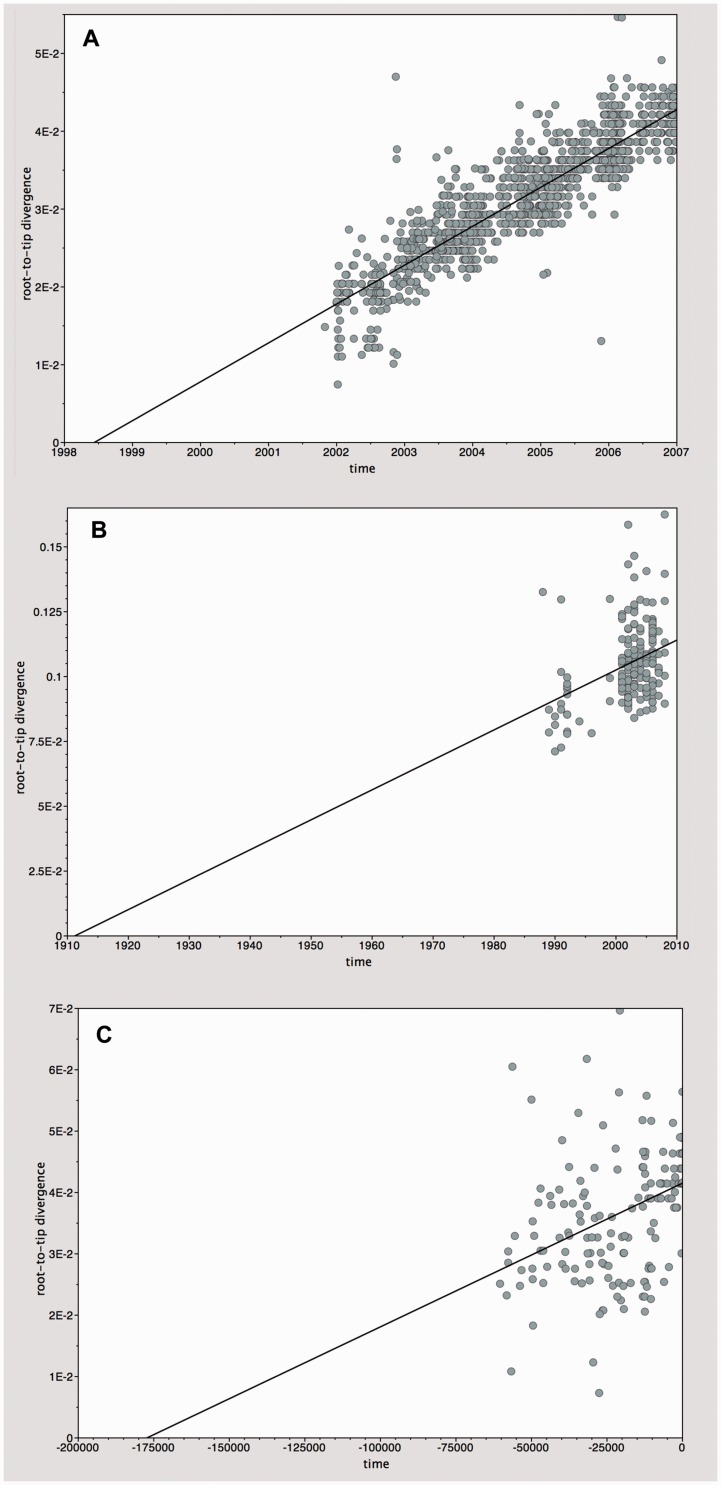
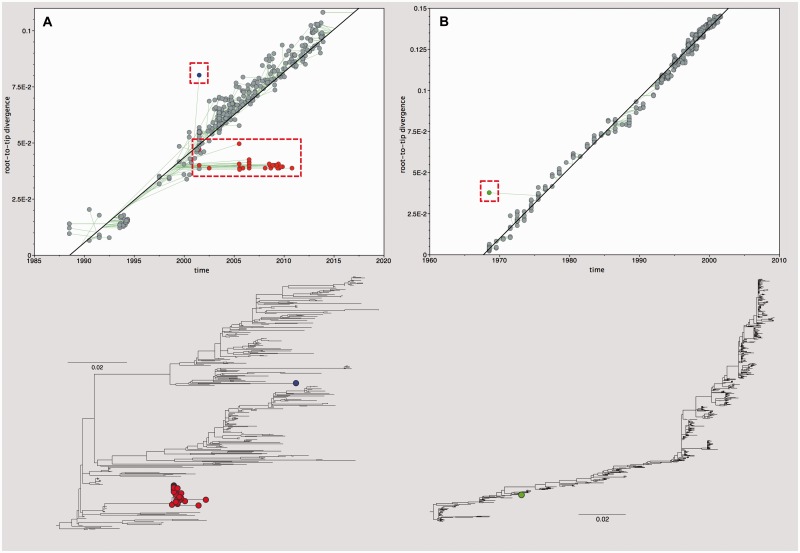
Gene sequences sampled at different points in time can be used to infer molecular phylogenies on a natural timescale of months or years, provided that the sequences in question undergo measurable amounts of evolutionary change between sampling times. Data sets with this property are termed heterochronous and have become increasingly common in several fields of biology, most notably the molecular epidemiology of rapidly evolving viruses. Here we introduce the cross-platform software tool, TempEst (formerly known as Path-O-Gen), for the visualization and analysis of temporally sampled sequence data. Given a molecular phylogeny and the dates of sampling for each sequence, TempEst uses an interactive regression approach to explore the association between genetic divergence through time and sampling dates. TempEst can be used to (1) assess whether there is sufficient temporal signal in the data to proceed with phylogenetic molecular clock analysis, and (2) identify sequences whose genetic divergence and sampling date are incongruent. Examination of the latter can help identify data quality problems, including errors in data annotation, sample contamination, sequence recombination, or alignment error. We recommend that all users of the molecular clock models implemented in BEAST first check their data using TempEst prior to analysis.
Keywords: evolutionary rate; model selection; molecular clock; phylogeny; regression.
Figures

References
-
- Buonagurio D. A., et al. (1986) ‘Evolution of Human Influenza A Viruses over 50 Years: Rapid, Uniform Rate of Change in NS Gene’, Science, 232: 980–2. - PubMed
-
- Drummond A. J., et al. (2003a) ‘Measurably Evolving Populations’, Trends in Ecology and Evolution, 18: 481–8.
-
- Drummond A. J., Pybus O. G., Rambaut A. (2003b) ‘Inference of Viral Evolutionary Rates from Molecular Sequences’, Advances in Parasitology, 54: 331–58. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
