

Identification of a novel GLA mutation (Y88C) in a Korean family with Fabry nephropathy: a case report
- PMID: 27776503
- PMCID: PMC5078899
- DOI: 10.1186/s12881-016-0338-7
Identification of a novel GLA mutation (Y88C) in a Korean family with Fabry nephropathy: a case report
Abstract
Background: Fabry disease is a rare X-linked lysosomal storage disorder caused by α-galactosidase A deficiency. With the advancement of molecular diagnostic tools, more disease-causing mutations in α-galactosidase A (GLA) have been identified in Fabry disease. We found a novel mutation in a Korean family with predominant renal manifestations of the disease.
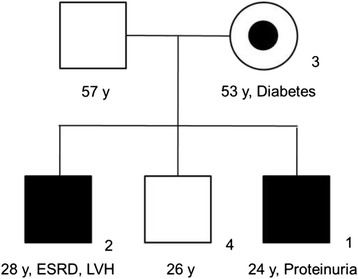
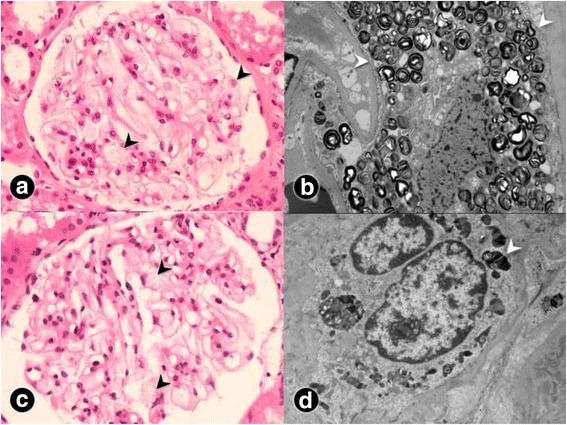
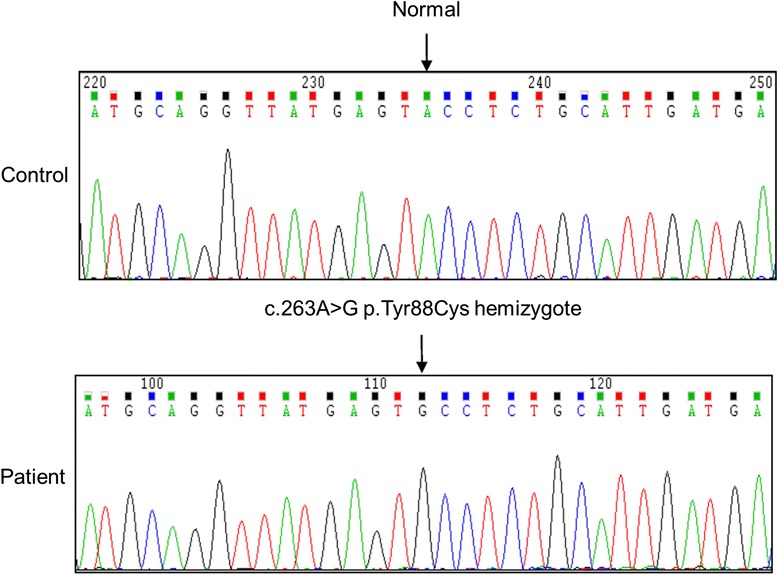
Case presentation: A 24-year-old man who wanted to donate a kidney to his 28-year-old brother with end-stage renal disease of unknown cause was evaluated. The 24-year-old man underwent percutaneous renal biopsy because of an accidentally found proteinuria. Electron microscopy of his renal biopsy showed numerous electron-dense multi-lamellar inclusions in the epithelial cytoplasm, typical for Fabry disease. Clinical and laboratory evaluation including the assessment of GLA enzyme activity and direct DNA sequencing in four members of the family were performed. Renal biopsy findings in the two affected male patients were described. Re-evaluation of a renal biopsy specimen of his 28-year-old brother obtained when he was diagnosed with renal failure revealed a very focal area of suspicious multilamellated structures in the Bowman's space. DNA sequencing on the young man, his brother, and his mother revealed a novel GLA gene mutation, c.263A > G (p.Tyr88Cys). The three all showed decreased α-galactosidase A activity.
Conclusion: A novel GLA mutation, c.263A > G (p.Tyr88Cys), was found in a Korean family with predominant renal manifestations of Fabry disease.
Keywords: Dialysis; Fabry disease; Kidney biopsy; Proteinuria; α-galactosidase A.
Figures
Similar articles
-
Functional Characterization and Pharmacological Evaluation of a Novel GLA Missense Mutation Found in a Severely Affected Fabry Disease Family.Nephron. 2020;144(3):147-155. doi: 10.1159/000503998. Epub 2019 Oct 30. Nephron. 2020. PMID: 31665721
-
A nonsense mutation (R220X) in the alpha-galactosidase A gene causes typical Fabry disease in both genders.Clin Nephrol. 2004 Mar;61(3):185-90. doi: 10.5414/cnp61185. Clin Nephrol. 2004. PMID: 15077869 Review.
-
Identification of a Novel GLA Gene Mutation, p.Ile239Met, in Fabry Disease With a Predominant Cardiac Phenotype.Int Heart J. 2017 May 31;58(3):454-458. doi: 10.1536/ihj.16-361. Epub 2017 May 12. Int Heart J. 2017. PMID: 28496025
-
The Identification of a Novel Pathogenic Variant of the GLA Gene Associated with a Classic Phenotype of Anderson-Fabry Disease: A Clinical and Molecular Study.Int J Mol Sci. 2025 Jan 8;26(2):470. doi: 10.3390/ijms26020470. Int J Mol Sci. 2025. PMID: 39859185 Free PMC article.
-
Diagnosing Fabry nephropathy: the challenge of multiple kidney disease.BMC Nephrol. 2023 Nov 21;24(1):344. doi: 10.1186/s12882-023-03388-8. BMC Nephrol. 2023. PMID: 37990184 Free PMC article. Review.
Cited by
-
Genotype⁻Phenotype Correlation in a New Fabry-Disease-Causing Mutation.Medicina (Kaunas). 2019 May 7;55(5):122. doi: 10.3390/medicina55050122. Medicina (Kaunas). 2019. PMID: 31067829 Free PMC article.
-
GLA missense and promoter variants co-segregating in a Chinese family with Fabry disease.Ann Transl Med. 2020 Jul;8(14):865. doi: 10.21037/atm-19-4510. Ann Transl Med. 2020. PMID: 32793709 Free PMC article.
-
Modeling of Fabry disease nephropathy using patient derived human induced pluripotent stem cells and kidney organoid system.J Transl Med. 2023 Feb 22;21(1):138. doi: 10.1186/s12967-023-03992-0. J Transl Med. 2023. PMID: 36814269 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
