

Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia
- PMID: 27777238
- PMCID: PMC5234216
- DOI: 10.1182/blood-2016-05-707653
Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia
Abstract
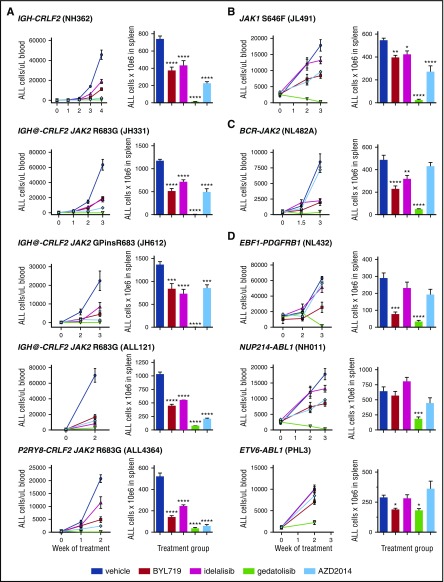
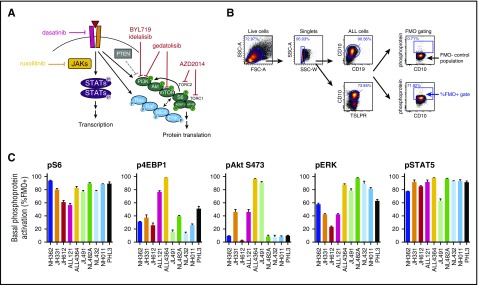
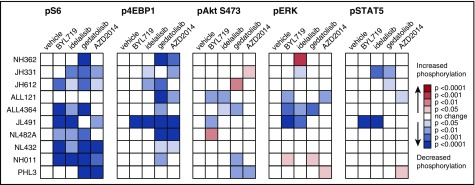
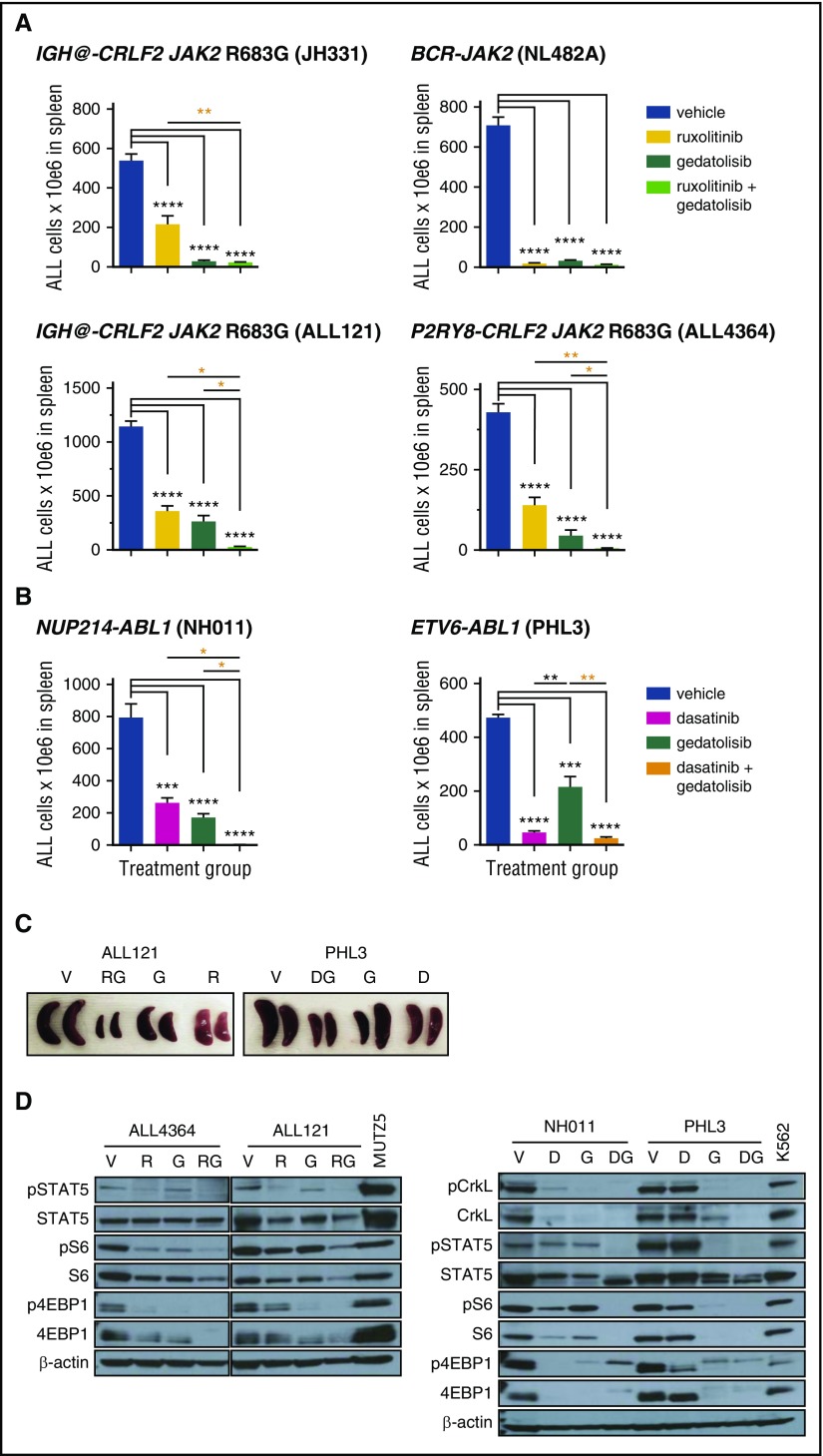
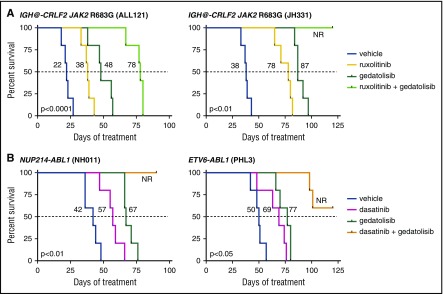
Philadelphia chromosome (Ph)-like B-cell acute lymphoblastic leukemia (Ph-like ALL) is associated with activated JAK/STAT, Abelson kinase (ABL), and/or phosphatidylinositol 3-kinase (PI3K) signaling and poor clinical outcomes. PI3K pathway signaling inhibitors have been minimally investigated in Ph-like ALL. We hypothesized that targeted inhibition of PI3Kα, PI3Kδ, PI3K/mTOR, or target of rapamycin complex 1/2 (TORC1/TORC2) would decrease leukemia proliferation and abrogate aberrant kinase signaling and that combined PI3K pathway and JAK inhibition or PI3K pathway and SRC/ABL inhibition would have superior efficacy compared to inhibitor monotherapy. We treated 10 childhood ALL patient-derived xenograft models harboring various Ph-like genomic alterations with 4 discrete PI3K pathway protein inhibitors and observed marked leukemia reduction and in vivo signaling inhibition in all models. Treatment with dual PI3K/mTOR inhibitor gedatolisib resulted in near eradication of ALL in cytokine receptor-like factor 2 (CRLF2)/JAK-mutant models with mean 92.2% (range, 86.0%-99.4%) reduction vs vehicle controls (P < .0001) and in prolonged animal survival. Gedatolisib also inhibited ALL proliferation in ABL/platelet-derived growth factor receptor (PDGFR)-mutant models with mean 66.9% (range, 42.0%-87.6%) reduction vs vehicle (P < .0001). Combined gedatolisib and ruxolitinib treatment of CRLF2/JAK-mutant models more effectively inhibited ALL proliferation than either inhibitor alone (P < .001) and further enhanced survival. Similarly, superior efficacy of combined gedatolisib and dasatinib was observed in ABL/PDGFR-mutant models (P < .001). Overall, PI3K/mTOR inhibition potently decreased ALL burden in vivo; antileukemia activity was further enhanced with combination inhibitor therapy. Clinical trials testing combinations of kinase inhibitors in Ph-like ALL patients are indicated.
© 2017 by The American Society of Hematology.
Figures
Comment in
-
One size doesn't fit all in Ph-like ALL.Blood. 2017 Jan 12;129(2):140-141. doi: 10.1182/blood-2016-11-749168. Blood. 2017. PMID: 28082291 Free PMC article.
References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57-70. - PubMed
-
- Teitell MA, Pandolfi PP. Molecular genetics of acute lymphoblastic leukemia. Annu Rev Pathol. 2009;4:175-198. - PubMed
-
- Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371(9617):1030-1043. - PubMed
-
- Jeha S, Pui CH. Risk-adapted treatment of pediatric acute lymphoblastic leukemia. Hematol Oncol Clin North Am. 2009;23(5):973-990. - PubMed
MeSH terms
Substances
Grants and funding
- I01 BX000918/BX/BLRD VA/United States
- P30 CA118100/CA/NCI NIH HHS/United States
- K08 CA184418/CA/NCI NIH HHS/United States
- U10 CA098413/CA/NCI NIH HHS/United States
- F31 HL124977/HL/NHLBI NIH HHS/United States
- U24 CA114766/CA/NCI NIH HHS/United States
- K12 CA076931/CA/NCI NIH HHS/United States
- U01 CA157937/CA/NCI NIH HHS/United States
- U10 CA098543/CA/NCI NIH HHS/United States
- U10 CA180899/CA/NCI NIH HHS/United States
- U10 CA180886/CA/NCI NIH HHS/United States
- T32 CA128583/CA/NCI NIH HHS/United States
- UL1 RR024134/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
