

Global DNA methylation profiling reveals new insights into epigenetically deregulated protein coding and long noncoding RNAs in CLL
- PMID: 27777635
- PMCID: PMC5062931
- DOI: 10.1186/s13148-016-0274-6
Global DNA methylation profiling reveals new insights into epigenetically deregulated protein coding and long noncoding RNAs in CLL
Abstract
Background: Methyl-CpG-binding domain protein enriched genome-wide sequencing (MBD-Seq) is a robust and powerful method for analyzing methylated CpG-rich regions with complete genome-wide coverage. In chronic lymphocytic leukemia (CLL), the role of CpG methylated regions associated with transcribed long noncoding RNAs (lncRNA) and repetitive genomic elements are poorly understood. Based on MBD-Seq, we characterized the global methylation profile of high CpG-rich regions in different CLL prognostic subgroups based on IGHV mutational status.
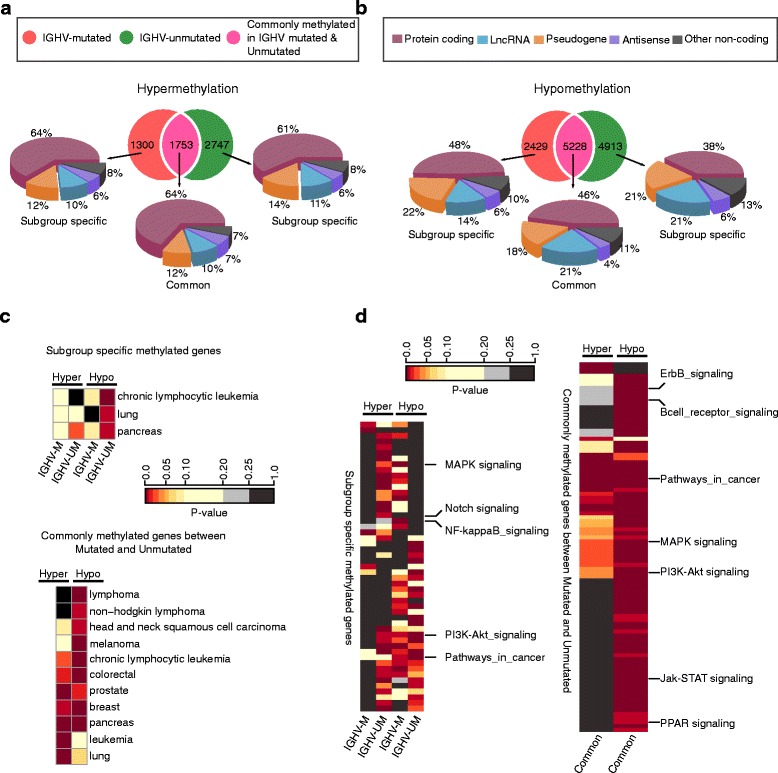
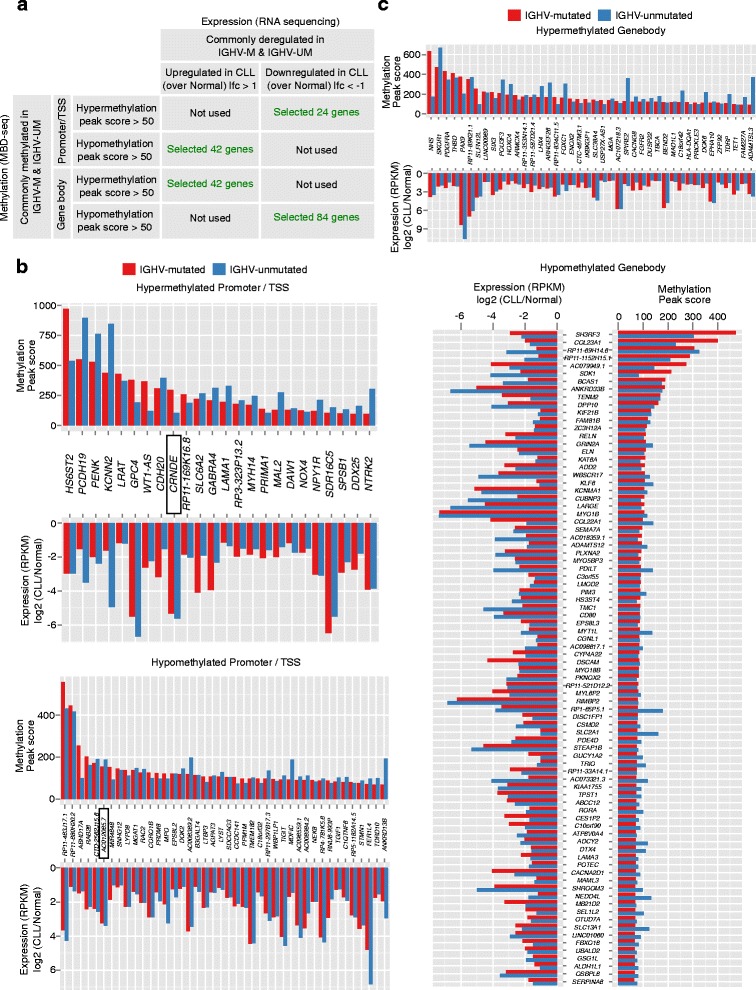
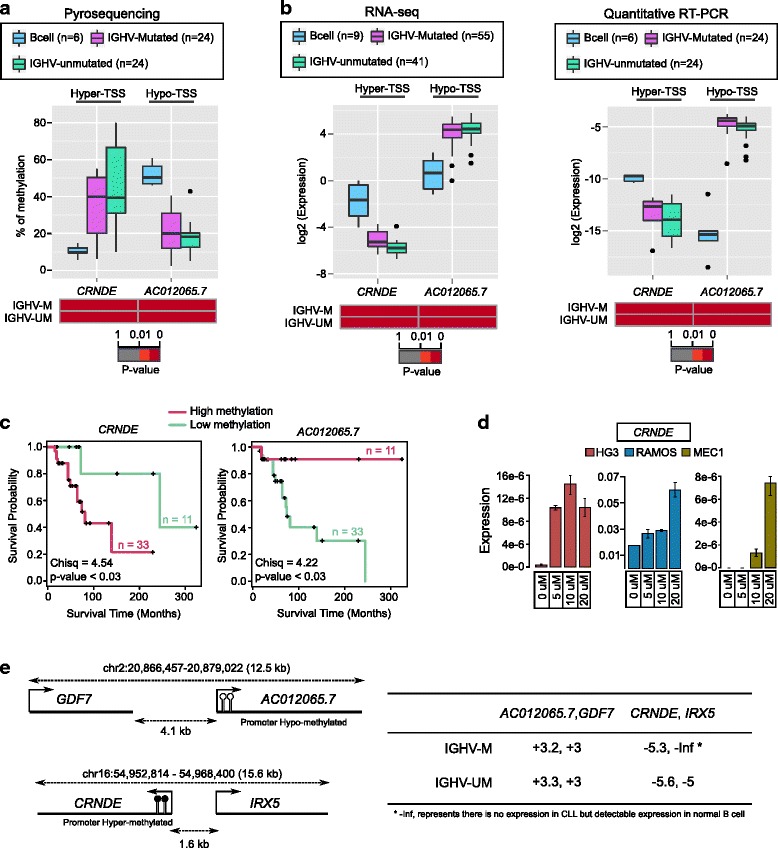
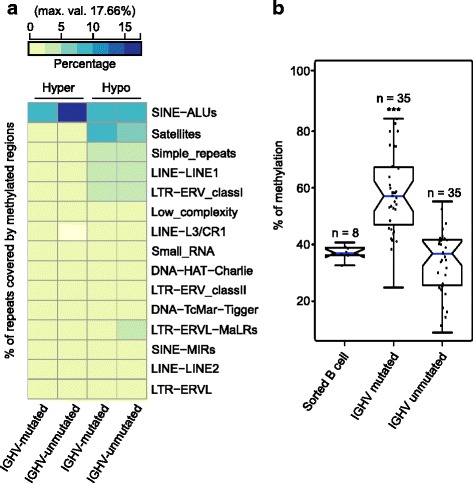
Results: Our study identified 5800 hypermethylated and 12,570 hypomethylated CLL-specific differentially methylated genes (cllDMGs) compared to normal controls. From cllDMGs, 40 % of hypermethylated and 60 % of hypomethylated genes were mapped to noncoding RNAs. In addition, we found that the major repetitive elements such as short interspersed elements (SINE) and long interspersed elements (LINE) have a high percentage of cllDMRs (differentially methylated regions) in IGHV subgroups compared to normal controls. Finally, two novel lncRNAs (hypermethylated CRNDE and hypomethylated AC012065.7) were validated in an independent CLL sample cohort (48 samples) compared with 6 normal sorted B cell samples using quantitative pyrosequencing analysis. The methylation levels showed an inverse correlation to gene expression levels analyzed by real-time quantitative PCR. Notably, survival analysis revealed that hypermethylation of CRNDE and hypomethylation of AC012065.7 correlated with an inferior outcome.
Conclusions: Thus, our comprehensive methylation analysis by MBD-Seq provided novel hyper and hypomethylated long noncoding RNAs, repetitive elements, along with protein coding genes as potential epigenetic-based CLL-signature genes involved in disease pathogenesis and prognosis.
Keywords: Chronic lymphocytic leukemia; DNA methylation; Hyper/hypomethylated regions; Repetitive elements and noncoding RNAs.
Figures

References
-
- Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, Buchbinder A, Budman D, Dittmar K, Kolitz J, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840–7. - PubMed
-
- Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V (H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–54. - PubMed
-
- Halldorsdottir AM, Kanduri M, Marincevic M, Mansouri L, Isaksson A, Goransson H, Axelsson T, Agarwal P, Jernberg-Wiklund H, Stamatopoulos K, et al. Mantle cell lymphoma displays a homogenous methylation profile: a comparative analysis with chronic lymphocytic leukemia. Am J Hematol. 2012;87:361–7. doi: 10.1002/ajh.23115. - DOI - PubMed
-
- Cahill N, Bergh AC, Kanduri M, Goransson-Kultima H, Mansouri L, Isaksson A, Ryan F, Smedby KE, Juliusson G, Sundstrom C, et al. 450 K-array analysis of chronic lymphocytic leukemia cells reveals global DNA methylation to be relatively stable over time and similar in resting and proliferative compartments. Leuk Off J Leuk Soc Am Leuk Res Fund UK. 2013;27:150–8. doi: 10.1038/leu.2012.245. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
