

Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome
- PMID: 27777904
- PMCID: PMC5073158
- DOI: 10.6065/apem.2016.21.3.126
Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome
Abstract
The Prader-Willi syndrome (PWS) is a human imprinting disorder resulting from genomic alterations that inactivate imprinted, paternally expressed genes in human chromosome region 15q11-q13. This genetic condition appears to be a contiguous gene syndrome caused by the loss of at least 2 of a number of genes expressed exclusively from the paternal allele, including SNRPN, MKRN3, MAGEL2, NDN and several snoRNAs, but it is not yet well known which specific genes in this region are associated with this syndrome. Prader-Will-Like syndrome (PWLS) share features of the PWS phenotype and the gene functions disrupted in PWLS are likely to lie in genetic pathways that are important for the development of PWS phenotype. However, the genetic basis of these rare disorders differs and the absence of a correct diagnosis may worsen the prognosis of these individuals due to the endocrine-metabolic malfunctioning associated with the PWS. Therefore, clinicians face a challenge in determining when to request the specific molecular test used to identify patients with classical PWS because the signs and symptoms of PWS are common to other syndromes such as PWLS. This review aims to provide an overview of current knowledge relating to the genetics of PWS and PWLS, with an emphasis on identification of patients that may benefit from further investigation and genetic screening.
Keywords: Genetic screening; Imprinting disorder; Prader-Willi syndrome; Prader-Willi-like syndrome.
Conflict of interest statement
No potential conflict of interest relevant to this article was reported.
Figures
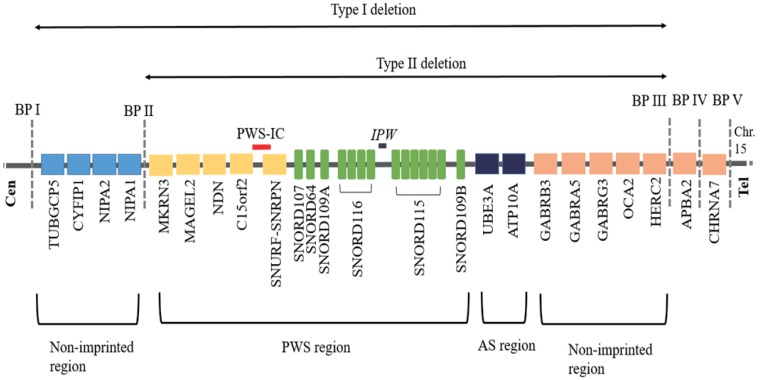
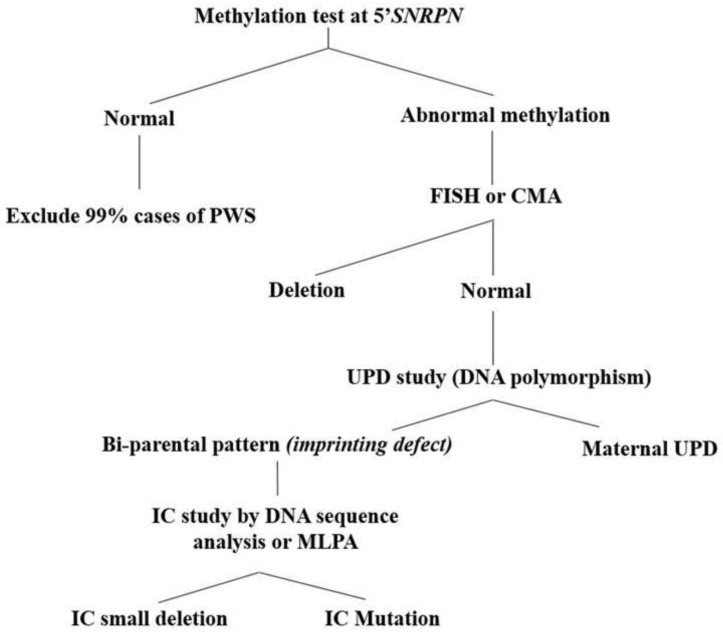
References
-
- Murrell A. Cross-talk between imprinted loci in Prader-Willi syndrome. Nat Genet. 2014;46:528–530. - PubMed
-
- Holland AJ, Treasure J, Coskeran P, Dallow J. Characteristics of the eating disorder in Prader-Willi syndrome: implications for treatment. J Intellect Disabil Res. 1995;39(Pt 5):373–381. - PubMed
-
- Swaab DF. Prader-Willi syndrome and the hypothalamus. Acta Paediatr Suppl. 1997;423:50–54. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
